Viewed and Counted at the Microscope Or Images Were Obtained
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Rac/Cdc42 Exchange Factor Complex Promotes Formation of Lateral filopodia and Blood Vessel Lumen Morphogenesis
ARTICLE Received 1 Oct 2014 | Accepted 26 Apr 2015 | Published 1 Jul 2015 DOI: 10.1038/ncomms8286 OPEN A Rac/Cdc42 exchange factor complex promotes formation of lateral filopodia and blood vessel lumen morphogenesis Sabu Abraham1,w,*, Margherita Scarcia2,w,*, Richard D. Bagshaw3,w,*, Kathryn McMahon2,w, Gary Grant2, Tracey Harvey2,w, Maggie Yeo1, Filomena O.G. Esteves2, Helene H. Thygesen2,w, Pamela F. Jones4, Valerie Speirs2, Andrew M. Hanby2, Peter J. Selby2, Mihaela Lorger2, T. Neil Dear4,w, Tony Pawson3,z, Christopher J. Marshall1 & Georgia Mavria2 During angiogenesis, Rho-GTPases influence endothelial cell migration and cell–cell adhesion; however it is not known whether they control formation of vessel lumens, which are essential for blood flow. Here, using an organotypic system that recapitulates distinct stages of VEGF-dependent angiogenesis, we show that lumen formation requires early cytoskeletal remodelling and lateral cell–cell contacts, mediated through the RAC1 guanine nucleotide exchange factor (GEF) DOCK4 (dedicator of cytokinesis 4). DOCK4 signalling is necessary for lateral filopodial protrusions and tubule remodelling prior to lumen formation, whereas proximal, tip filopodia persist in the absence of DOCK4. VEGF-dependent Rac activation via DOCK4 is necessary for CDC42 activation to signal filopodia formation and depends on the activation of RHOG through the RHOG GEF, SGEF. VEGF promotes interaction of DOCK4 with the CDC42 GEF DOCK9. These studies identify a novel Rho-family GTPase activation cascade for the formation of endothelial cell filopodial protrusions necessary for tubule remodelling, thereby influencing subsequent stages of lumen morphogenesis. 1 Institute of Cancer Research, Division of Cancer Biology, 237 Fulham Road, London SW3 6JB, UK. -

Double Inhibition and Activation Mechanisms of Ephexin Family Rhogefs
Double inhibition and activation mechanisms of Ephexin family RhoGEFs Meng Zhanga,1, Lin Linb,1, Chao Wanga,2, and Jinwei Zhub,2b,,2 aMinistry of Education Key Laboratory for Membraneless Organelles and Cellular Dynamics, Hefei National Laboratory for Physical Sciences at the Microscale, School of Life Sciences, Division of Life Sciences and Medicine, University of Science and Technology of China, 230027 Hefei, China; and bBio-X Institutes, Key Laboratory for the Genetics of Developmental and Neuropsychiatric Disorders, Ministry of Education, Shanghai Jiao Tong University, Shanghai 200240, China Edited by Alfred Wittinghofer, Max Planck Institute of Molecular Physiology-Dortmund, Dortmund, Germany, and accepted by Editorial Board Member Brenda A. Schulman January 20, 2021 (received for review December 2, 2020) Ephexin family guanine nucleotide exchange factors (GEFs) trans- functions of Ephexin2 and Ephexin3 remain elusive although fer signals from Eph tyrosine kinase receptors to Rho GTPases, they are known to activate RhoA (8). Therefore, the Ephexin which play critical roles in diverse cellular processes, as well as family RhoGEFs serve as the regulatory hubs that link Ephrin- cancers and brain disorders. Here, we elucidate the molecular basis Eph signaling with cytoskeletal dynamics through spatiotemporal underlying inhibition and activation of Ephexin family RhoGEFs. regulation of Rho GTPases. Dysfunctions of Eph-Ephexin–mediated The crystal structures of partially and fully autoinhibited Ephexin4 Rho signaling have been associated with a variety of diseases, ranging reveal that the complete autoinhibition requires both N- and from cancers to brain disorders (18–22). C-terminal inhibitory modes, which can operate independently to Each member of the Ephexin family proteins contains a Dbl impede Ras homolog family member G (RhoG) access. -

A Rhog-Mediated Signaling Pathway That Modulates Invadopodia Dynamics in Breast Cancer Cells Silvia M
© 2017. Published by The Company of Biologists Ltd | Journal of Cell Science (2017) 130, 1064-1077 doi:10.1242/jcs.195552 RESEARCH ARTICLE A RhoG-mediated signaling pathway that modulates invadopodia dynamics in breast cancer cells Silvia M. Goicoechea, Ashtyn Zinn, Sahezeel S. Awadia, Kyle Snyder and Rafael Garcia-Mata* ABSTRACT micropinocytosis, bacterial uptake, phagocytosis and leukocyte One of the hallmarks of cancer is the ability of tumor cells to invade trans-endothelial migration (deBakker et al., 2004; Ellerbroek et al., surrounding tissues and metastasize. During metastasis, cancer cells 2004; Jackson et al., 2015; Katoh et al., 2006, 2000; van Buul et al., degrade the extracellular matrix, which acts as a physical barrier, by 2007). Recent studies have revealed that RhoG plays a role in tumor developing specialized actin-rich membrane protrusion structures cell invasion and may contribute to the formation of invadopodia called invadopodia. The formation of invadopodia is regulated by Rho (Hiramoto-Yamaki et al., 2010; Kwiatkowska et al., 2012). GTPases, a family of proteins that regulates the actin cytoskeleton. Invadopodia are actin-rich adhesive structures that form in the Here, we describe a novel role for RhoG in the regulation of ventral surface of cancer cells and allow them to degrade the invadopodia disassembly in human breast cancer cells. Our results extracellular matrix (ECM) (Gimona et al., 2008). Formation of show that RhoG and Rac1 have independent and opposite roles invadopodia involves a series of steps that include the disassembly in the regulation of invadopodia dynamics. We also show that SGEF of focal adhesions and stress fibers, and the relocalization of several (also known as ARHGEF26) is the exchange factor responsible of their components into the newly formed invadopodia (Hoshino for the activation of RhoG during invadopodia disassembly. -

Endothelial Barrier Function and Leukocyte Transmigration in Atherosclerosis
biomedicines Review Endothelial Barrier Function and Leukocyte Transmigration in Atherosclerosis Thijs J. Sluiter 1,2 , Jaap D. van Buul 3 , Stephan Huveneers 4, Paul H. A. Quax 1,2 and Margreet R. de Vries 1,2,* 1 Department of Vascular Surgery, Leiden University Medical Center, 2333 ZA Leiden, The Netherlands; [email protected] (T.J.S.); [email protected] (P.H.A.Q.) 2 Einthoven Laboratory for Experimental Vascular Medicine, Leiden University Medical Center, 2333 ZA Leiden, The Netherlands 3 Sanquin Research and Landsteiner Laboratory, Leeuwenhoek Centre for Advanced Microscopy, Swammerdam Institute for Life Sciences, University of Amsterdam, 1066 CX Amsterdam, The Netherlands; [email protected] 4 Department of Medical Biochemistry, Amsterdam Cardiovascular Sciences, Amsterdam University Medical Center, Location AMC, University of Amsterdam, 1105 AZ Amsterdam, The Netherlands; [email protected] * Correspondence: [email protected]; Tel.: +31-(71)-526-5147 Abstract: The vascular endothelium is a highly specialized barrier that controls passage of fluids and migration of cells from the lumen into the vessel wall. Endothelial cells assist leukocytes to extravasate and despite the variety in the specific mechanisms utilized by different leukocytes to cross different vascular beds, there is a general principle of capture, rolling, slow rolling, arrest, crawling, and ultimately diapedesis via a paracellular or transcellular route. In atherosclerosis, the barrier function of the endothelium is impaired leading to uncontrolled leukocyte extravasation and Citation: Sluiter, T.J.; van Buul, J.D.; vascular leakage. This is also observed in the neovessels that grow into the atherosclerotic plaque Huveneers, S.; Quax, P.H.A.; de Vries, leading to intraplaque hemorrhage and plaque destabilization. -

ICAM-1 (Phospho Tyr512) Polyclonal Antibody
ICAM-1 Monoclonal Antibody Catalog No : YM1051 Reactivity : Human Applications : WB,IF/ICC Gene Name : ICAM1 Protein Name : Intercellular adhesion molecule 1 Human Gene Id : 3383 Human Swiss Prot P05362 No : Mouse Swiss Prot P13597 No : Immunogen : Purified recombinant human ICAM-1 (N-terminus) protein fragments expressed in E.coli. Specificity : ICAM-1 Monoclonal Antibody detects endogenous levels of ICAM-1 protein. Formulation : Purified mouse monoclonal in buffer containing 0.1M Tris-Glycine (pH 7.4, 150 mM NaCl) with 0.2% sodium azide, 50% glycerol. Source : Mouse Dilution : Western Blot: 1/1000 - 1/2000. Immunofluorescence: 1/100 - 1/500. Not yet tested in other applications. Purification : Affinity purification Concentration : 1 mg/ml Storage Stability : -20°C/1 year Cell Pathway : Cell adhesion molecules (CAMs),Natural killer cell mediated cytotoxicity,Leukocyte transendothelial migration,Viral myocarditis, 1 / 2 Background : intercellular adhesion molecule 1(ICAM1) Homo sapiens This gene encodes a cell surface glycoprotein which is typically expressed on endothelial cells and cells of the immune system. It binds to integrins of type CD11a / CD18, or CD11b / CD18 and is also exploited by Rhinovirus as a receptor. [provided by RefSeq, Jul 2008], Function : function:ICAM proteins are ligands for the leukocyte adhesion protein LFA-1 (integrin alpha-L/beta-2). During leukocyte trans-endothelial migration, ICAM1 engagement promotes the assembly of endothelial apical cups through SGEF and RHOG activation. In case of rhinovirus infection acts as a cellular receptor for the virus.,online information:ICAM-1,online information:Icosahedral capsid structure,online information:Intercellular adhesion molecule entry,polymorphism:Homozygotes with ICAM1-Kalifi Met-56 seem to have an increased risk for cerebral malaria.,PTM:Monoubiquitinated, which is promoted by MARCH9 and leads to endocytosis.,similarity:Belongs to the immunoglobulin superfamily. -
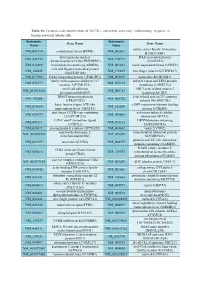
Systematic Name Gene Name Systematic Name Gene Name NM 001710 Complement Factor B(CFB) NM 052831 Solute Carrier Family 18 Member
Table S1: Genome-wide identification of SGLT2i`s interaction with early inflammatory response in human proximal tubular cells. Systematic Systematic Gene Name Gene Name Name Name solute carrier family 18 member NM_001710 complement factor B(CFB) NM_052831 B1(SLC18B1) heterogeneous nuclear DAZ associated protein NM_031372 NM_170711 ribonucleoprotein D like(HNRNPDL) 1(DAZAP1) NM_014299 bromodomain containing 4(BRD4) NM_001261 cyclin dependent kinase 9(CDK9) cilia and flagella associated protein NM_182628 NM_178835 zinc finger protein 827(ZNF827) 100(CFAP100) NM_017906 PAK1 interacting protein 1(PAK1IP1) NM_024015 homeobox B4(HOXB4) family with sequence similarity 167 ankyrin repeat and LEM domain NM_053279 NM_015114 member A(FAM167A) containing 2(ANKLE2) small cell adhesion ARP3 actin related protein 3 NM_001031628 NM_005721 glycoprotein(SMAGP) homolog(ACTR3) TRAF3 interacting protein actin related protein 2/3 complex NM_147686 NM_005720 2(TRAF3IP2) subunit 1B(ARPC1B) basic leucine zipper ATF-like cAMP responsive element binding NM_018664 NM_182898 transcription factor 3(BATF3) protein 5(CREB5) zinc finger CCCH-type containing activation induced cytidine NM_025079 NM_020661 12A(ZC3H12A) deaminase(AICDA) C-X-C motif chemokine ligand DENN domain containing NM_001511 NM_015213 1(CXCL1) 5A(DENND5A) NM_025072 prostaglandin E synthase 2(PTGES2) NM_004665 vanin 2(VNN2) superoxide dismutase 2, mitochondrial ribosomal protein NM_001024465 NM_016070 mitochondrial(SOD2) S23(MRPS23) jumonji and AT-rich interaction NM_033199 urocortin 2(UCN2) NM_004973 -

The Human Papillomavirus E6 PDZ Binding Motif: from Life Cycle to Malignancy
Viruses 2015, 7, 3530-3551; doi:10.3390/v7072785 OPEN ACCESS viruses ISSN 1999-4915 www.mdpi.com/journal/viruses Review The Human Papillomavirus E6 PDZ Binding Motif: From Life Cycle to Malignancy Ketaki Ganti, Justyna Broniarczyk, Wiem Manoubi, Paola Massimi, Suruchi Mittal, David Pim, Anita Szalmas, Jayashree Thatte, Miranda Thomas, Vjekoslav Tomaic´ and Lawrence Banks * International Center for Genetic Engineering and Biotechnology, Area Science Park, Padriciano 99, Trieste 34149, Italy; E-Mails: [email protected] (K.G.); [email protected] (J.B.); [email protected] (W.M.); [email protected] (P.M.); [email protected] (S.M.); [email protected] (D.P.); [email protected] (A.S.); [email protected] (J.T.); [email protected] (M.T.); [email protected] (V.T.) * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +39-040-375-7328. Academic Editor: Joanna Parish Received: 6 May 2015 / Accepted: 25 June 2015 / Published: 2 July 2015 Abstract: Cancer-causing HPV E6 oncoproteins are characterized by the presence of a PDZ binding motif (PBM) at their extreme carboxy terminus. It was long thought that this region of E6 had a sole function to confer interaction with a defined set of cellular substrates. However, more recent studies have shown that the E6 PBM has a complex pattern of regulation, whereby phosphorylation within the PBM can regulate interaction with two classes of cellular proteins: those containing PDZ domains and the members of the 14-3-3 family of proteins. In this review, we explore the roles that the PBM and its ligands play in the virus life cycle, and subsequently how these can inadvertently contribute towards the development of malignancy. -

Supplemental Data Heidel Et Al
Supplemental data Heidel et al. Table of Contents 1. Sequencing strategy and statistics ...................................................................................................... 2 2. Genome structure ............................................................................................................................... 2 2.1 Extrachromosal elements .............................................................................................................. 2 2.2 Chromosome structure ................................................................................................................. 3 2.3 Repetitive elements ...................................................................................................................... 5 3. Coding sequences ................................................................................................................................ 5 3.1 Homopolymer tracts ..................................................................................................................... 5 3.2 Gene families and orthology relationships ................................................................................... 7 3.3 Synteny analysis .......................................................................................................................... 11 4. Protein functional domains ............................................................................................................... 12 5. Protein families ................................................................................................................................. -

Identification of Novel Regulatory Mechanisms for Cdc42 Gtpase-Activating Protein Cdgap/ARHGAP31, a Protein Involved in Development and Cancer
Identification of novel regulatory mechanisms for Cdc42 GTPase-activating protein CdGAP/ARHGAP31, a protein involved in development and cancer Ali Ben Djoudi Ouadda Department of Anatomy & Cell Biology McGill University, Montréal, Québec, Canada Submitted October, 2016 A thesis submitted to McGill University in partial fulfillment of the requirements of the degree of Doctor of Philosophy © Ali Ben Djoudi Ouadda, 2016 Acknowledgments I would like to express my deepest thanks and appreciation to my supervisor, Dr Nathalie Lamarche-Vane, who has opened the door of her laboratory and gave me the opportunity to pursue an excellent research training. Without her kindness, support, guidance and persistent help this thesis would not have been possible. I would like to thank my mentor Dr. Carlos Morales, who has supported, encouraged and guided me with valuable advice from the beginning. I would like also to thank my advisory committee members, Dr. Isabelle Rouiller and Dr. Peter Siegel for their encouragement and precious scientific inputs and feedbacks. I would like to acknowledge the Fonds de Recherche du Québec-Santé (FRSQ) which awarded me a Doctoral Training Scholarship and the McGill Faculty of Medicine/Department of Anatomy & Cell Biology which granted me a Doctoral Internal Scholarship, GREAT Travel and Merit Awards. In addition, a thank you to my colleagues in the Department of Anatomy & Cell Biology, RI-MUHC, IRCM and IRIC for their help and collaboration, either with reagents or scientific discussion and troubleshooting. Special thanks to Martin, Yi and Vilayphone for their precious help and support during my early days in the lab, and to Philippe, Sadig, Fereshteh, Hidetaka, Tristan, Jonathan and Judith for their help, kindness and availability. -

Identification of 15 Novel Risk Loci for Coronary Artery Disease and Genetic
www.nature.com/scientificreports OPEN Identification of 15 novel risk loci for coronary artery disease and genetic risk of recurrent events, Received: 27 January 2017 Accepted: 21 April 2017 atrial fibrillation and heart failure Published: xx xx xxxx Niek Verweij 1, Ruben N. Eppinga1, Yanick Hagemeijer 1 & Pim van der Harst1,2,3 Coronary artery disease (CAD) is the major cause of morbidity and mortality in the world. Identification of novel genetic determinants may provide new opportunities for developing innovative strategies to predict, prevent and treat CAD. Therefore, we meta-analyzed independent genetic variants passing P <× 10−5 in CARDIoGRAMplusC4D with novel data made available by UK Biobank. Of the 161 genetic variants studied, 71 reached genome wide significance (p < 5 × 10−8) including 15 novel loci. These novel loci include multiple genes that are involved in angiogenesis (TGFB1, ITGB5, CDH13 and RHOA) and 2 independent variants in the TGFB1 locus. We also identifiedSGEF as a candidate gene in one of the novel CAD loci. SGEF was previously suggested as a therapeutic target based on mouse studies. The genetic risk score of CAD predicted recurrent CAD events and cardiovascular mortality. We also identified significant genetic correlations between CAD and other cardiovascular conditions, including heart failure and atrial fibrillation. In conclusion, we substantially increased the number of loci convincingly associated with CAD and provide additional biological and clinical insights. Coronary artery disease (CAD) is a major burden of morbidity and mortality to Western society1. CAD is driven by a complex interplay of multiple genetic and environmental factors that jointly give rise to a plethora of molec- ular interactions resulting in a complex and heterogeneous phenotype. -

SGEF Is Overexpressedin Gliolastoma and Mediates TWEAK-Fn14
THE TWEAK-FN14 LIGAND RECEPTOR AXIS PROMOTES GLIOBLASTOMA CELL INVASION AND SURVIVAL VIA ACTIVATION OF MULTIPLE GEF-RHO GTPASE SIGNALING SYSTEMS Item Type text; Electronic Thesis Authors Fortin Ensign, Shannon Patricia Publisher The University of Arizona. Rights Copyright © is held by the author. Digital access to this material is made possible by the College of Medicine - Phoenix, University of Arizona. Further transmission, reproduction or presentation (such as public display or performance) of protected items is prohibited except with permission of the author. Download date 25/09/2021 12:31:38 Link to Item http://hdl.handle.net/10150/528171 SGEF is overexpressed in glioblastoma and mediates TWEAK‐Fn14‐induced cell survival Shannon P. Fortin Ensign1,2, Ian T. Mathews,2 Harshil Dhruv2, Marc H. Symons3, Jann N. Sarkaria4, Nhan L. Tran2 1University of Arizona, Phoenix & Tucson, AZ; 2Translational Genomics Research Institute, Phoenix, AZ; 3The Feinstein Institute for Medical Research at North Shore‐LIJ, Manhasset, NY; 4Mayo Clinic, Rochester, MN Abstract Glioblastoma (GB) is the highest grade and most common form of primary adult brain tumors. Despite surgical removal followed by concomitant radiation and chemotherapy with the alkylating agent temozolomide (TMZ), GB tumors develop treatment resistance and ultimately recur. Impaired response to treatment occurs Figure 7. SGEF mRNA expression is inducible via TWEAK cytokine stimulation. T98G glioma cells rapidly conferring a median survival of just fifteen months. Thus, it were cultured in reduced serum (0.5% FBS DMEM) for 16 hours prior to stimulation with TWEAK (100ng/mL) for the indicated times. SGEF mRNA (A) and protein (B) expression were analyzed via is necessary to identify the genetic and signaling mechanisms that qPCR with fold change relative to histone and via western blotting with the indicated antibodies, promote tumor resistance in order to develop targeted therapies to respectively. -

The EGFR/Mir-338-3P/EYA2 Axis Controls Breast Tumor Growth and Lung Metastasis
Citation: Cell Death and Disease (2017) 8, e2928; doi:10.1038/cddis.2017.325 OPEN Official journal of the Cell Death Differentiation Association www.nature.com/cddis The EGFR/miR-338-3p/EYA2 axis controls breast tumor growth and lung metastasis Yingchun Liang1,8, Xiaojie Xu1,8, Tao Wang2,8, Ying Li1,3, Wenye You1,3, Jing Fu1, Yang Liu1,4, Shuai Jin1,4, Quanbo Ji1,5, Wei Zhao6, Qi Song3, Ling Li1, Tian Hong1, Junjian Huang1, Zhaohui Lyu*,7 and Qinong Ye*,1 Dysregulation of the epidermal growth factor receptor (EGFR) promotes cancer cell growth, invasion and metastasis. However, its relevant downstream effectors are still limited. Here, we show that EGFR promotes breast tumor growth and metastasis by downregulating the tumor suppressor micoRNA-338-3p (miR-338-3p) and activating the EYA2 (EYA transcriptional coactivator and phosphatase 2) oncoprotein. EGFR represses miR-338-3p expression largely through HIF1α transcription factor. miR-338-3p inhibits EYA2expression by binding to the 3′-untranslated region of EYA2. EGFR increases EYA2 expression via HIF1α repression of miR-338- 3p. Through the miR-338-3p/EYA2pathway, EGFR increases breast cancer cell growth, epithelial-to-mesenchymal transition, migration, invasion and lung metastasis in vitro and in a allograft tumor mouse model in vivo. In breast cancer patients, miR-338-3p expression negatively correlates with the expression of EGFR and EYA2, EGFR status positively associates with EYA2 expression, and miR-338-3p and EYA2 predict breast cancer lung metastasis when expressed in primary breast cancers. These data suggest that the miR-338-3p/ EYA2 axis contributes to EGFR-mediated tumor growth and lung metastasis and that miR-338-3p activation or EYA2 inhibition or combination therapy targeting EGFR/miR-338-3p/EYA2 axis may be a promising way to treat patients with metastatic cancer.