Biol 200 – Professor Richard Roy
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Computation and Visualization of Protein Topology Graphs Including Ligand Information
Computation and Visualization of Protein Topology Graphs Including Ligand Information Tim Schäfer1, Patrick May2, and Ina Koch1 1 Institute of Computer Science, Department of Molecular Bioinformatics, Johann Wolfgang Goethe-University Frankfurt (Main), Robert-Mayer-Straße 11–15, 60325 Frankfurt (Main), Germany, [email protected] 2 Luxembourg Centre for Systems Biomedicine, University of Luxembourg, Campus Belval, 7 Avenue des Hauts-Fourneaux, L–4362 Esch-sur-Alzette, Luxembourg Abstract Motivation: Ligand information is of great interest to understand protein function. Protein structure topology can be modeled as a graph with secondary structure elements as vertices and spatial contacts between them as edges. Meaningful representations of such graphs in 2D are required for the visual inspection, comparison and analysis of protein folds, but their automatic visualization is still challenging. We present an approach which solves this task, supports different graph types and can optionally include ligand contacts. Results: Our method extends the field of protein structure description and visualization by including ligand information. It generates a mathematically unique representation and high- quality 2D plots of the secondary structure of a protein based on a protein-ligand graph. This graph is computed from 3D atom coordinates in PDB files and the corresponding SSE assignments of the DSSP algorithm. The related software supports different notations and allows a rapid visualization of protein structures. It can also export graphs in various standard file formats so they can be used with other software. Our approach visualizes ligands in relationship to protein structure topology and thus represents a useful tool for exploring protein structures. Availability: The software is released under an open source license and available at http://www.bioinformatik.uni-frankfurt.de/ in the Software section under Visualization of Protein Ligand Graphs. -

Predictive Energy Landscapes for Folding Α-Helical Transmembrane Proteins
Predictive energy landscapes for folding α-helical transmembrane proteins Bobby L. Kim, Nicholas P. Schafer, and Peter G. Wolynes1 Departments of Chemistry and Physics and Astronomy and the Center for Theoretical Biological Physics, Rice University, Houston, TX 77005 Contributed by Peter G. Wolynes, June 11, 2014 (sent for review May 19, 2014; reviewed by Zaida A. Luthey-Schulten, Shoji Takada, and Margaret Cheung) We explore the hypothesis that the folding landscapes of mem- folding. Starting with Khorana’swork(7),numerousα-helical brane proteins are funneled once the proteins’ topology within the transmembrane proteins have been refolded from a chemically membrane is established. We extend a protein folding model, denatured state in vitro (8). This indicates that at least some the associative memory, water-mediated, structure, and energy transmembrane domains may not require the translocon to fold model (AWSEM) by adding an implicit membrane potential and properly. In addition, recent experiments on a few α-helical reoptimizing the force field to account for the differing nature of transmembrane proteins have succeeded in characterizing the the interactions that stabilize proteins within lipid membranes, structure of transition state ensembles, in a manner like that used yielding a model that we call AWSEM-membrane. Once the pro- for globular proteins. These studies suggest that native contacts tein topology is set in the membrane, hydrophobic attractions are important in the folding nucleus but may not represent the play a lesser role in finding the native structure, whereas po- whole story (9, 10). Whether membrane proteins possess energy lar–polar attractions are more important than for globular pro- landscapes as funneled as globular proteins remains an open teins. -

Protein Folding and the Organization of the Protein Topology Universe
Opinion TRENDS in Biochemical Sciences Vol.30 No.1 January 2005 Protein folding and the organization of the protein topology universe Kresten Lindorff-Larsen1, Peter Røgen2, Emanuele Paci3, Michele Vendruscolo1 and Christopher M. Dobson1 1University of Cambridge, Department of Chemistry, Lensfield Road, Cambridge, UK, CB2 1EW 2Department of Mathematics, Technical University of Denmark, Building 303, DK-2800 Kongens Lyngby, Denmark 3University of Zu¨ rich, Department of Biochemistry, Winterthurerstrasse 190, 8057 Zu¨ rich, Switzerland The mechanism by which proteins fold to their native ensembles has shown that establishing the correct overall states has been the focus of intense research in recent topology of the polypeptide chain is a crucial aspect of years. The rate-limiting event in the folding reaction is protein folding. This observation is in accord with a series the formation of a conformation in a set known as the of studies that have shown that the folding rate of a transition-state ensemble. The structural features pre- protein, to a first approximation, can be related to the sent within such ensembles have now been analysed for entropic cost of forming the native-like topology [16–22]. a series of proteins using data from a combination of The structural changes occurring during protein fold- biochemical and biophysical experiments together with ing have also been analysed in detail for a series of computer-simulation methods. These studies show that proteins and we discuss some of these studies here. The the topology of the transition state is determined by a results enable the topological view of folding to be set of interactions involving a small number of key reconciled with the well-established concept of nucleation residues and, in addition, that the topology of the [23] by showing that – despite the many different ways in transition state is closer to that of the native state than which a given topology could, in principle, be generated – to that of any other fold in the protein universe. -

Using the Spycatcher-Spytag and Related Systems for Labeling and Localizing Bacterial Proteins
International Journal of Molecular Sciences Review Catching a SPY: Using the SpyCatcher-SpyTag and Related Systems for Labeling and Localizing Bacterial Proteins Daniel Hatlem , Thomas Trunk, Dirk Linke and Jack C. Leo * Bacterial Cell Surface Group, Section for Evolution and Genetics, Department of Biosciences, University of Oslo, 0316 Oslo, Norway; [email protected] (D.H.); [email protected] (T.T.); [email protected] (D.L.) * Correspondence: [email protected]; Tel.: +47-228-59027 Received: 2 April 2019; Accepted: 26 April 2019; Published: 30 April 2019 Abstract: The SpyCatcher-SpyTag system was developed seven years ago as a method for protein ligation. It is based on a modified domain from a Streptococcus pyogenes surface protein (SpyCatcher), which recognizes a cognate 13-amino-acid peptide (SpyTag). Upon recognition, the two form a covalent isopeptide bond between the side chains of a lysine in SpyCatcher and an aspartate in SpyTag. This technology has been used, among other applications, to create covalently stabilized multi-protein complexes, for modular vaccine production, and to label proteins (e.g., for microscopy). The SpyTag system is versatile as the tag is a short, unfolded peptide that can be genetically fused to exposed positions in target proteins; similarly, SpyCatcher can be fused to reporter proteins such as GFP, and to epitope or purification tags. Additionally, an orthogonal system called SnoopTag-SnoopCatcher has been developed from an S. pneumoniae pilin that can be combined with SpyCatcher-SpyTag to produce protein fusions with multiple components. Furthermore, tripartite applications have been produced from both systems allowing the fusion of two peptides by a separate, catalytically active protein unit, SpyLigase or SnoopLigase. -

UC San Diego UC San Diego Electronic Theses and Dissertations
UC San Diego UC San Diego Electronic Theses and Dissertations Title Structural analysis of a forkhead-associated domain from the type III secretion system protein YscD Permalink https://escholarship.org/uc/item/6410g6n9 Author Gamez, Alicia Margarita Publication Date 2011 Peer reviewed|Thesis/dissertation eScholarship.org Powered by the California Digital Library University of California UNIVERSITY OF CALIFORNIA, SAN DIEGO Structural analysis of a forkhead-associated domain from the type III secretion system protein YscD A dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy in Chemistry by Alicia Margarita Gamez Committee in charge: Professor Partho Ghosh, Chair Professor Michael Burkart Professor Daniel Donoghue Professor Victor Nizet Professor Susan Taylor 2011 The Dissertation of Alicia Margarita Gamez is approved, and it is acceptable in quality and form for publication on microfilm and electronically: Chair University of California, San Diego 2011 iii DEDICATION This thesis is dedicated to my family. iv TABLE OF CONTENTS Signature Page .......................................................................................................... iii Dedication ................................................................................................................. iv Table of Contents ....................................................................................................... v List of Figures .......................................................................................................... -

APEX2 Proximity Proteomics Resolves Flagellum Subdomains and Identifies Flagellum Tip-Specific
bioRxiv preprint doi: https://doi.org/10.1101/2020.03.09.984815; this version posted March 12, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 APEX2 proximity proteomics resolves flagellum subdomains and identifies flagellum tip-specific 2 proteins in Trypanosoma brucei 3 4 Daniel E. Vélez-Ramírez,a,b Michelle M. Shimogawa,a Sunayan Ray,a* Andrew Lopez,a Shima 5 Rayatpisheh,c* Gerasimos Langousis,a* Marcus Gallagher-Jones,d Samuel Dean,e James A. Wohlschlegel,c 6 Kent L. Hill,a,f,g# 7 8 aDepartment of Microbiology, Immunology, and Molecular Genetics, University of California Los 9 Angeles, Los Angeles, California, USA 10 bPosgrado en Ciencias Biológicas, Universidad Nacional Autónoma de México, Coyoacán, Ciudad de 11 México, México 12 cDepartment of Biological Chemistry, University of California Los Angeles, Los Angeles, California, USA 13 dDepartment of Chemistry and Biochemistry, UCLA-DOE Institute for Genomics and Proteomics, Los 14 Angeles, California, USA 15 eWarwick Medical School, University of Warwick, Coventry, England, United Kingdom 16 fMolecular Biology Institute, University of California Los Angeles, Los Angeles, California, USA 17 gCalifornia NanoSystems Institute, University of California Los Angeles, Los Angeles, California, USA 18 19 Running title: APEX2 proteomics in Trypanosoma brucei 20 21 #Address correspondence to Kent L. Hill, [email protected]. Page 1 of 33 bioRxiv preprint doi: https://doi.org/10.1101/2020.03.09.984815; this version posted March 12, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. -

Machine Learning Methods for Protein Structure Prediction Jianlin Cheng, Allison N
IEEE REVIEWS IN BIOMEDICAL ENGINEERING, VOL. 1, 2008 41 Machine Learning Methods for Protein Structure Prediction Jianlin Cheng, Allison N. Tegge, Member, IEEE, and Pierre Baldi, Senior Member, IEEE Methodological Review Abstract—Machine learning methods are widely used in bioin- formatics and computational and systems biology. Here, we review the development of machine learning methods for protein structure prediction, one of the most fundamental problems in structural biology and bioinformatics. Protein structure predic- tion is such a complex problem that it is often decomposed and attacked at four different levels: 1-D prediction of structural features along the primary sequence of amino acids; 2-D predic- tion of spatial relationships between amino acids; 3-D prediction Fig. 1. Protein sequence-structure-function relationship. A protein is a linear of the tertiary structure of a protein; and 4-D prediction of the polypeptide chain composed of 20 different kinds of amino acids represented quaternary structure of a multiprotein complex. A diverse set of by a sequence of letters (left). It folds into a tertiary (3-D) structure (middle) both supervised and unsupervised machine learning methods has composed of three kinds of local secondary structure elements (helix – red; beta- strand – yellow; loop – green). The protein with its native 3-D structure can carry been applied over the years to tackle these problems and has sig- out several biological functions in the cell (right). nificantly contributed to advancing the state-of-the-art of protein structure prediction. In this paper, we review the development and application of hidden Markov models, neural networks, support vector machines, Bayesian methods, and clustering methods in structure elements are further packed to form a tertiary structure 1-D, 2-D, 3-D, and 4-D protein structure predictions. -

The Relationship Between APOL1 Structure and Function: Clinical Implications
Kidney360 Publish Ahead of Print, published on November 4, 2020 as doi:10.34067/KID.0002482020 The relationship between APOL1 structure and function: Clinical implications Sethu M. Madhavan1, Matthias Buck2 1Department of Medicine, The Ohio State University, Columbus, OH, 2Department of Physiology & Biophysics, Case Western Reserve University School of Medicine, Cleveland, OH Corresponding author: Sethu M. Madhavan, M.D. 495 W 12th Avenue Columbus, OH 43210 Email: [email protected] Phone: 614-293-6389 1 Copyright 2020 by American Society of Nephrology. Abstract Common variants in the APOL1 gene are associated with an increased risk of nondiabetic kidney disease in individuals of African ancestry. Mechanisms by which APOL1 variants mediate kidney disease pathogenesis are not well understood. Amino acid changes resulting from the kidney disease-associated APOL1 variants alter the three-dimensional structure and conformational dynamics of the C-terminal α- helical domain of the protein, which can rationalize the functional consequences. Understanding the three-dimensional structure of the protein with and without the risk variants can provide insights into the pathogenesis of kidney diseases mediated by APOL1 variants. Introduction Population-based studies have established a strong association of two variants in the APOL1 gene with the excess risk of nondiabetic chronic kidney disease (CKD) in individuals of African ancestry.1-5 One variant involves the substitution of two amino acids (S342G and I384M; termed G1), and the other the deletion of two consecutive amino acids (N388 and Y389; termed G2) compared to the ancestral nonrisk allele termed G0. APOL1 G1 and G2 variants are common in individuals with African ancestry, with at least 50 % of individuals carrying one copy of the risk allele and 15 % with two copies of the risk allele.1,3 Despite the strong association of APOL1 variants with kidney disease, the molecular mechanisms by which these APOL1 variants contribute to CKD pathogenesis and progression remains unclear. -

Predicting Protein Secondary and Supersecondary Structure
29 Predicting Protein Secondary and Supersecondary Structure 29.1 Introduction............................................ 29-1 Background • Difficulty of general protein structure prediction • A bottom-up approach 29.2 Secondary structure ................................... 29-5 Early approaches • Incorporating local dependencies • Exploiting evolutionary information • Recent developments and conclusions 29.3 Tight turns ............................................. 29-13 29.4 Beta hairpins........................................... 29-15 29.5 Coiled coils ............................................. 29-16 Early approaches • Incorporating local dependencies • Predicting oligomerization • Structure-based predictions • Predicting coiled-coil protein interactions Mona Singh • Promising future directions Princeton University 29.6 Conclusions ............................................ 29-23 29.1 Introduction Proteins play a key role in almost all biological processes. They take part in, for example, maintaining the structural integrity of the cell, transport and storage of small molecules, catalysis, regulation, signaling and the immune system. Linear protein molecules fold up into specific three-dimensional structures, and their functional properties depend intricately upon their structures. As a result, there has been much effort, both experimental and computational, in determining protein structures. Protein structures are determined experimentally using either x-ray crystallography or nuclear magnetic resonance (NMR) spectroscopy. While -

The Development of the Prediction of Protein Structure
6 The Development of the Prediction of Protein Structure Gerald D. Fasman I. Introduction .................................................................... 194 II. Protein Topology. .. 196 III. Techniques of Protein Prediction ................................................... 198 A. Sequence Alignment .......................................................... 199 B. Hydrophobicity .............................................................. 200 C. Minimum Energy Calculations ................................................. 202 IV. Approaches to Protein Conformation ................................................ 203 A. Solvent Accessibility ......................................................... 203 B. Packing of Residues .......................................................... 204 C. Distance Geometry ........................................................... 205 D. Amino Acid Physicochemical Properties ......................................... 205 V. Prediction of the Secondary Structure of Proteins: a Helix, ~ Strands, and ~ Turn .......... 208 A. ~ Turns .................................................................... 209 B. Evaluation of Predictive Methodologies .......................................... 218 C. Other Predictive Algorithms ................................................... 222 D. Chou-Fasman Algorithm ...................................................... 224 E. Class Prediction ............................................................. 233 VI. Prediction of Tertiary Structure ................................................... -
Protein Topology Function
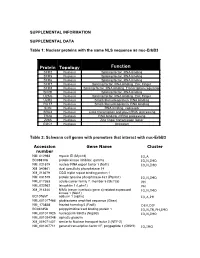
SUPPLEMENTAL INFORMATION SUPPLEMENTAL DATA Table 1: Nuclear proteins with the same NLS sequence as nuc-ErbB3 Protein Topology Function SFR2 Nucleus Splicing factor, RNA binding SFR5 Nucleus Splicing factor, RNA binding SFR6 Nucleus Splicing factor, RNA binding SFR7 Nucleus Splicing factor, RNA binding, Zinc Finger SFR8 Nucleus Splicing factor, RNA binding, Transcription regulation SFRB Nucleus Splicing factor, RNA binding U2AG Nucleus Splicing factor, RNA binding, Zinc Finger U2R2 Nucleus Small ribonucleoprotein, RNA binding RU17 Nucleus Small ribonucleoprotein, RNA binding SON Nucleus RNA binding, repressor SRA4 Nucleus Links transcription and pre-mRNA processing TR2A Nucleus RNA binding, mRNA processing Z265 Nucleus Zinc finger transcription factor GGC1 Nucleus Unknown Table 2: Schwann cell genes with promoters that interact with nuc-ErbB3 Accession Gene Name Cluster number NM_012983 myosin ID (Myo1d) IO,A BC088108 protein kinase inhibitor, gamma IO,N,IMO NM_021579 nuclear RNA export factor 1 (Nxf1) IO,N,IMO XM_340861 dual specificity phosphatase 14 XM_213679 CGG triplet repeat binding protein 1 NM_031579 protein tyrosine phosphatase 4a1 (Ptp4a1) IO,N,IMO NM_017353 solute carrier family 7, member 5 (Slc7a5) PH NM_022962 latrophilin 1 (Lphn1) PH XM_214340 NIMA (never in mitosis gene a)-related expressed IO,N,IMO kinase 1 (Nek1) BC107657 adducin 1 (alpha) IO,A,PH NM_001017486 glioblastoma amplified sequence (Gbas) NM_173838 frizzled homolog 5 (Fzd5) DEV,DIF BC061858 polypyrimidine tract binding protein 1 IO,N,TR,PH,IMO NM_001011925 nucleoporin -

Ÿþf R O N T M a T T
Nediljko Budisa Engineering the Genetic Code Engineering the Genetic Code. Nediljko Budisa Copyright 8 2006 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim ISBN: 3-527-31243-9 Related Titles K. H. Nierhaus, D. N. Wilson (eds.) S. Brakmann, K. Johnsson (eds.) Protein Synthesis and Ribosome Directed Molecular Evolution of Structure Proteins Translating the Genome or How to Improve Enzymes for Biocatalysis 597 pages with approx. 150 figures and approx. 40 368 pages with 121 figures and 15 tables tables 2002 2004 Hardcover Hardcover ISBN 3-527-30423-1 ISBN 3-527-30638-2 G. Walsh S. Brakmann, A. Schwienhorst (eds.) Proteins Evolutionary Methods in Biochemistry and Biotechnology Biotechnology 560 pages Clever Tricks for Directed Evolution 2002 227 pages with 115 figures and 11 tables Softcover 2004 ISBN 0-471-89907-0 Hardcover ISBN 3-527-30799-0 N. Sewald, H.-D. Jakubke Peptides: Chemistry and Biology 597 pages with 163 figures and 33 tables 2002 Hardcover ISBN 3-527-30405-3 Nediljko Budisa Engineering the Genetic Code Expanding the Amino Acid Repertoire for the Design of Novel Proteins Nediljko Budisa 9 All books published by Wiley-VCH are carefully Max-Planck-Institute of Biochemistry produced. Nevertheless, authors, editors, and Division of Bioorganic Chemistry publisher do not warrant the information Am Klopferspitz 18A contained in these books, including this book, 82152 Martinsried to be free of errors. Readers are advised to keep Germany in mind that statements, data, illustrations, procedural details or other items may inadvertently be inaccurate. Library of Congress Card No.: applied for British Library Cataloguing-in-Publication Data A catalogue record for this book is available from the British Library.