Data Formats Document Table of Contents
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplemental Note Hominoid Fission of Chromosome 14/15 and Role Of
Supplemental Note Supplemental Note Hominoid fission of chromosome 14/15 and role of segmental duplications Giuliana Giannuzzi, Michele Pazienza, John Huddleston, Francesca Antonacci, Maika Malig, Laura Vives, Evan E. Eichler and Mario Ventura 1. Analysis of the macaque contig spanning the hominoid 14/15 fission site We grouped contig clones based on their FISH pattern on human and macaque chromosomes (Groups F1, F2, F3, G1, G2, G3, and G4). Group F2 clones (Hsa15b- and Hsa15c-positive) showed a single signal on macaque 7q and three signal clusters on human chromosome 15: at the orthologous 15q26, as expected, as well as at 15q11–14 and 15q24–25, which correspond to the actual and ancestral pericentromeric regions, respectively (Ventura et al. 2003). Indeed, this locus contains an LCR15 copy (Pujana et al. 2001) in both the macaque and human genomes. Most BAC clones were one-end anchored in the human genome (chr15:100,028–100,071 kb). In macaque this region experienced a 64 kb duplicative insertion from chromosome 17 (orthologous to human chromosome 13) (Figure 2), with the human configuration (absence of insertion) likely being the ancestral state because it is identical in orangutan and marmoset. Four Hsa15b- positive clones mapped on macaque and human chromosome 19, but neither human nor macaque assemblies report the STS Hsa15b duplicated at this locus. The presence of assembly gaps in macaque may explain why the STS is not annotated in this region. We aligned the sequence of macaque CH250-70H12 (AC187495.2) versus its human orthologous sequence (hg18 chr15:100,039k-100,170k) and found a 12 kb human expansion through tandem duplication of a ~100 bp unit corresponding to a portion of exon 20 of DNM1 (Figure S3). -

Genetic Variation Across the Human Olfactory Receptor Repertoire Alters Odor Perception
bioRxiv preprint doi: https://doi.org/10.1101/212431; this version posted November 1, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Genetic variation across the human olfactory receptor repertoire alters odor perception Casey Trimmer1,*, Andreas Keller2, Nicolle R. Murphy1, Lindsey L. Snyder1, Jason R. Willer3, Maira Nagai4,5, Nicholas Katsanis3, Leslie B. Vosshall2,6,7, Hiroaki Matsunami4,8, and Joel D. Mainland1,9 1Monell Chemical Senses Center, Philadelphia, Pennsylvania, USA 2Laboratory of Neurogenetics and Behavior, The Rockefeller University, New York, New York, USA 3Center for Human Disease Modeling, Duke University Medical Center, Durham, North Carolina, USA 4Department of Molecular Genetics and Microbiology, Duke University Medical Center, Durham, North Carolina, USA 5Department of Biochemistry, University of Sao Paulo, Sao Paulo, Brazil 6Howard Hughes Medical Institute, New York, New York, USA 7Kavli Neural Systems Institute, New York, New York, USA 8Department of Neurobiology and Duke Institute for Brain Sciences, Duke University Medical Center, Durham, North Carolina, USA 9Department of Neuroscience, University of Pennsylvania School of Medicine, Philadelphia, Pennsylvania, USA *[email protected] ABSTRACT The human olfactory receptor repertoire is characterized by an abundance of genetic variation that affects receptor response, but the perceptual effects of this variation are unclear. To address this issue, we sequenced the OR repertoire in 332 individuals and examined the relationship between genetic variation and 276 olfactory phenotypes, including the perceived intensity and pleasantness of 68 odorants at two concentrations, detection thresholds of three odorants, and general olfactory acuity. -

13 Genomics and Bioinformatics
Enderle / Introduction to Biomedical Engineering 2nd ed. Final Proof 5.2.2005 11:58am page 799 13 GENOMICS AND BIOINFORMATICS Spencer Muse, PhD Chapter Contents 13.1 Introduction 13.1.1 The Central Dogma: DNA to RNA to Protein 13.2 Core Laboratory Technologies 13.2.1 Gene Sequencing 13.2.2 Whole Genome Sequencing 13.2.3 Gene Expression 13.2.4 Polymorphisms 13.3 Core Bioinformatics Technologies 13.3.1 Genomics Databases 13.3.2 Sequence Alignment 13.3.3 Database Searching 13.3.4 Hidden Markov Models 13.3.5 Gene Prediction 13.3.6 Functional Annotation 13.3.7 Identifying Differentially Expressed Genes 13.3.8 Clustering Genes with Shared Expression Patterns 13.4 Conclusion Exercises Suggested Reading At the conclusion of this chapter, the reader will be able to: & Discuss the basic principles of molecular biology regarding genome science. & Describe the major types of data involved in genome projects, including technologies for collecting them. 799 Enderle / Introduction to Biomedical Engineering 2nd ed. Final Proof 5.2.2005 11:58am page 800 800 CHAPTER 13 GENOMICS AND BIOINFORMATICS & Describe practical applications and uses of genomic data. & Understand the major topics in the field of bioinformatics and DNA sequence analysis. & Use key bioinformatics databases and web resources. 13.1 INTRODUCTION In April 2003, sequencing of all three billion nucleotides in the human genome was declared complete. This landmark of modern science brought with it high hopes for the understanding and treatment of human genetic disorders. There is plenty of evidence to suggest that the hopes will become reality—1631 human genetic diseases are now associated with known DNA sequences, compared to the less than 100 that were known at the initiation of the Human Genome Project (HGP) in 1990. -

Genomics and Its Impact on Science and Society: the Human Genome Project and Beyond
DOE/SC-0083 Genomics and Its Impact on Science and Society The Human Genome Project and Beyond U.S. Department of Energy Genome Research Programs: genomics.energy.gov A Primer ells are the fundamental working units of every living system. All the instructions Cneeded to direct their activities are contained within the chemical DNA (deoxyribonucleic acid). DNA from all organisms is made up of the same chemical and physical components. The DNA sequence is the particular side-by-side arrangement of bases along the DNA strand (e.g., ATTCCGGA). This order spells out the exact instruc- tions required to create a particular organism with protein complex its own unique traits. The genome is an organism’s complete set of DNA. Genomes vary widely in size: The smallest known genome for a free-living organism (a bac- terium) contains about 600,000 DNA base pairs, while human and mouse genomes have some From Genes to Proteins 3 billion (see p. 3). Except for mature red blood cells, all human cells contain a complete genome. Although genes get a lot of attention, the proteins DNA in each human cell is packaged into 46 chro- perform most life functions and even comprise the mosomes arranged into 23 pairs. Each chromosome is majority of cellular structures. Proteins are large, complex a physically separate molecule of DNA that ranges in molecules made up of chains of small chemical com- length from about 50 million to 250 million base pairs. pounds called amino acids. Chemical properties that A few types of major chromosomal abnormalities, distinguish the 20 different amino acids cause the including missing or extra copies or gross breaks and protein chains to fold up into specific three-dimensional rejoinings (translocations), can be detected by micro- structures that define their particular functions in the cell. -

Genetic Effects on Microsatellite Diversity in Wild Emmer Wheat (Triticum Dicoccoides) at the Yehudiyya Microsite, Israel
Heredity (2003) 90, 150–156 & 2003 Nature Publishing Group All rights reserved 0018-067X/03 $25.00 www.nature.com/hdy Genetic effects on microsatellite diversity in wild emmer wheat (Triticum dicoccoides) at the Yehudiyya microsite, Israel Y-C Li1,3, T Fahima1,MSRo¨der2, VM Kirzhner1, A Beiles1, AB Korol1 and E Nevo1 1Institute of Evolution, University of Haifa, Mount Carmel, Haifa 31905, Israel; 2Institute for Plant Genetics and Crop Plant Research, Corrensstrasse 3, 06466 Gatersleben, Germany This study investigated allele size constraints and clustering, diversity. Genome B appeared to have a larger average and genetic effects on microsatellite (simple sequence repeat number (ARN), but lower variance in repeat number 2 repeat, SSR) diversity at 28 loci comprising seven types of (sARN), and smaller number of alleles per locus than genome tandem repeated dinucleotide motifs in a natural population A. SSRs with compound motifs showed larger ARN than of wild emmer wheat, Triticum dicoccoides, from a shade vs those with perfect motifs. The effects of replication slippage sun microsite in Yehudiyya, northeast of the Sea of Galilee, and recombinational effects (eg, unequal crossing over) on Israel. It was found that allele distribution at SSR loci is SSR diversity varied with SSR motifs. Ecological stresses clustered and constrained with lower or higher boundary. (sun vs shade) may affect mutational mechanisms, influen- This may imply that SSR have functional significance and cing the level of SSR diversity by both processes. natural constraints. -

Gene Prediction and Genome Annotation
A Crash Course in Gene and Genome Annotation Lieven Sterck, Bioinformatics & Systems Biology VIB-UGent [email protected] ProCoGen Dissemination Workshop, Riga, 5 nov 2013 “Conifer sequencing: basic concepts in conifer genomics” “This Project is financially supported by the European Commission under the 7th Framework Programme” Genome annotation: finding the biological relevant features on a raw genomic sequence (in a high throughput manner) ProCoGen Dissemination Workshop, Riga, 5 nov 2013 Thx to: BSB - annotation team • Lieven Sterck (Ectocarpus, higher plants, conifers, … ) • Yao-cheng Lin (Fungi, conifers, …) • Stephane Rombauts (green alga, mites, …) • Bram Verhelst (green algae) • Pierre Rouzé • Yves Van de Peer ProCoGen Dissemination Workshop, Riga, 5 nov 2013 Annotation experience • Plant genomes : A.thaliana & relatives (e.g. A.lyrata), Poplar, Physcomitrella patens, Medicago, Tomato, Vitis, Apple, Eucalyptus, Zostera, Spruce, Oak, Orchids … • Fungal genomes: Laccaria bicolor, Melampsora laricis- populina, Heterobasidion, other basidiomycetes, Glomus intraradices, Pichia pastoris, Geotrichum Candidum, Candida ... • Algal genomes: Ostreococcus spp, Micromonas, Bathycoccus, Phaeodactylum (and other diatoms), E.hux, Ectocarpus, Amoebophrya … • Animal genomes: Tetranychus urticae, Brevipalpus spp (mites), ... ProCoGen Dissemination Workshop, Riga, 5 nov 2013 Why genome annotation? • Raw sequence data is not useful for most biologists • To be meaningful to them it has to be converted into biological significant knowledge -

Assembly and Annotation of an Ashkenazi Human Reference Genome
bioRxiv preprint doi: https://doi.org/10.1101/2020.03.18.997395; this version posted March 18, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Assembly and Annotation of an Ashkenazi Human Reference Genome Alaina Shumate1,2,† Aleksey V. Zimin1,2,† Rachel M. Sherman1,3 Daniela Puiu1,3 Justin M. Wagner4 Nathan D. Olson4 Mihaela Pertea1,2 Marc L. Salit5 Justin M. Zook4 Steven L. Salzberg1,2,3,6* 1Center for Computational Biology, Johns Hopkins University, Baltimore, MD 2Department of Biomedical Engineering, Johns Hopkins University, Baltimore, MD 3Department of Computer Science, Johns Hopkins University, Baltimore, MD 4National Institute of Standards and Technology, Gaithersburg, MD 5Joint Initiative for Metrology in Biology, Stanford University, Stanford, CA 6Department of Biostatistics, Johns Hopkins University, Baltimore, MD †These authors contributed equally to this work. *Corresponding author. Email: [email protected] Abstract Here we describe the assembly and annotation of the genome of an Ashkenazi individual and the creation of a new, population-specific human reference genome. This genome is more contiguous and more complete than GRCh38, the latest version of the human reference genome, and is annotated with highly similar gene content. The Ashkenazi reference genome, Ash1, contains 2,973,118,650 nucleotides as compared to 2,937,639,212 in GRCh38. Annotation identified 20,157 protein-coding genes, of which 19,563 are >99% identical to their counterparts on GRCh38. Most of the remaining genes have small differences. -

Small Variants Frequently Asked Questions (FAQ) Updated September 2011
Small Variants Frequently Asked Questions (FAQ) Updated September 2011 Summary Information for each Genome .......................................................................................................... 3 How does Complete Genomics map reads and call variations? ........................................................................... 3 How do I assess the quality of a genome produced by Complete Genomics?................................................ 4 What is the difference between “Gross mapping yield” and “Both arms mapped yield” in the summary file? ............................................................................................................................................................................. 5 What are the definitions for Fully Called, Partially Called, Half-Called and No-Called?............................ 5 In the summary-[ASM-ID].tsv file, how is the number of homozygous SNPs calculated? ......................... 5 In the summary-[ASM-ID].tsv file, how is the number of heterozygous SNPs calculated? ....................... 5 In the summary-[ASM-ID].tsv file, how is the total number of SNPs calculated? .......................................... 5 In the summary-[ASM-ID].tsv file, what regions of the genome are included in the “exome”? .............. 6 In the summary-[ASM-ID].tsv file, how is the number of SNPs in the exome calculated? ......................... 6 In the summary-[ASM-ID].tsv file, how are variations in potentially redundant regions of the genome counted? ..................................................................................................................................................................... -

Epigenetics Analysis and Integrated Analysis of Multiomics Data, Including Epigenetic Data, Using Artificial Intelligence in the Era of Precision Medicine
biomolecules Review Epigenetics Analysis and Integrated Analysis of Multiomics Data, Including Epigenetic Data, Using Artificial Intelligence in the Era of Precision Medicine Ryuji Hamamoto 1,2,*, Masaaki Komatsu 1,2, Ken Takasawa 1,2 , Ken Asada 1,2 and Syuzo Kaneko 1 1 Division of Molecular Modification and Cancer Biology, National Cancer Center Research Institute, 5-1-1 Tsukiji, Chuo-ku, Tokyo 104-0045, Japan; [email protected] (M.K.); [email protected] (K.T.); [email protected] (K.A.); [email protected] (S.K.) 2 Cancer Translational Research Team, RIKEN Center for Advanced Intelligence Project, 1-4-1 Nihonbashi, Chuo-ku, Tokyo 103-0027, Japan * Correspondence: [email protected]; Tel.: +81-3-3547-5271 Received: 1 December 2019; Accepted: 27 December 2019; Published: 30 December 2019 Abstract: To clarify the mechanisms of diseases, such as cancer, studies analyzing genetic mutations have been actively conducted for a long time, and a large number of achievements have already been reported. Indeed, genomic medicine is considered the core discipline of precision medicine, and currently, the clinical application of cutting-edge genomic medicine aimed at improving the prevention, diagnosis and treatment of a wide range of diseases is promoted. However, although the Human Genome Project was completed in 2003 and large-scale genetic analyses have since been accomplished worldwide with the development of next-generation sequencing (NGS), explaining the mechanism of disease onset only using genetic variation has been recognized as difficult. Meanwhile, the importance of epigenetics, which describes inheritance by mechanisms other than the genomic DNA sequence, has recently attracted attention, and, in particular, many studies have reported the involvement of epigenetic deregulation in human cancer. -

The Economic Impact and Functional Applications of Human Genetics and Genomics
The Economic Impact and Functional Applications of Human Genetics and Genomics Commissioned by the American Society of Human Genetics Produced by TEConomy Partners, LLC. Report Authors: Simon Tripp and Martin Grueber May 2021 TEConomy Partners, LLC (TEConomy) endeavors at all times to produce work of the highest quality, consistent with our contract commitments. However, because of the research and/or experimental nature of this work, the client undertakes the sole responsibility for the consequence of any use or misuse of, or inability to use, any information or result obtained from TEConomy, and TEConomy, its partners, or employees have no legal liability for the accuracy, adequacy, or efficacy thereof. Acknowledgements ASHG and the project authors wish to thank the following organizations for their generous support of this study. Invitae Corporation, San Francisco, CA Regeneron Pharmaceuticals, Inc., Tarrytown, NY The project authors express their sincere appreciation to the following indi- viduals who provided their advice and input to this project. ASHG Government and Public Advocacy Committee Lynn B. Jorde, PhD ASHG Government and Public Advocacy Committee (GPAC) Chair, President (2011) Professor and Chair of Human Genetics George and Dolores Eccles Institute of Human Genetics University of Utah School of Medicine Katrina Goddard, PhD ASHG GPAC Incoming Chair, Board of Directors (2018-2020) Distinguished Investigator, Associate Director, Science Programs Kaiser Permanente Northwest Melinda Aldrich, PhD, MPH Associate Professor, Department of Medicine, Division of Genetic Medicine Vanderbilt University Medical Center Wendy Chung, MD, PhD Professor of Pediatrics in Medicine and Director, Clinical Cancer Genetics Columbia University Mira Irons, MD Chief Health and Science Officer American Medical Association Peng Jin, PhD Professor and Chair, Department of Human Genetics Emory University Allison McCague, PhD Science Policy Analyst, Policy and Program Analysis Branch National Human Genome Research Institute Rebecca Meyer-Schuman, MS Human Genetics Ph.D. -
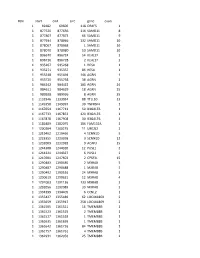
Chr Start End Size Gene Exon 1 69482 69600 118 OR4F5 1 1 877520
#chr start end size gene exon 1 69482 69600 118 OR4F5 1 1 877520 877636 116 SAMD11 8 1 877807 877873 66 SAMD11 9 1 877934 878066 132 SAMD11 10 1 878067 878068 1 SAMD11 10 1 878070 878080 10 SAMD11 10 1 896670 896724 54 KLHL17 2 1 896726 896728 2 KLHL17 2 1 935267 935268 1 HES4 1 1 935271 935357 86 HES4 1 1 955548 955694 146 AGRN 1 1 955720 955758 38 AGRN 1 1 984242 984422 180 AGRN 24 1 984611 984629 18 AGRN 25 1 989928 989936 8 AGRN 35 1 1132946 1133034 88 TTLL10 13 1 1149358 1149397 39 TNFRSF4 1 1 1167654 1167713 59 B3GALT6 1 1 1167733 1167853 120 B3GALT6 1 1 1167878 1167908 30 B3GALT6 1 1 1181889 1182075 186 FAM132A 1 1 1200204 1200215 11 UBE2J2 2 1 1219462 1219466 4 SCNN1D 5 1 1223355 1223358 3 SCNN1D 12 1 1232009 1232018 9 ACAP3 15 1 1244308 1244320 12 PUSL1 2 1 1244321 1244327 6 PUSL1 2 1 1247601 1247603 2 CPSF3L 15 1 1290483 1290485 2 MXRA8 5 1 1290487 1290488 1 MXRA8 5 1 1290492 1290516 24 MXRA8 5 1 1290619 1290631 12 MXRA8 4 1 1291003 1291136 133 MXRA8 3 1 1292056 1292089 33 MXRA8 2 1 1334399 1334405 6 CCNL2 1 1 1355427 1355489 62 LOC441869 2 1 1355659 1355917 258 LOC441869 2 1 1361505 1361521 16 TMEM88B 1 1 1361523 1361525 2 TMEM88B 1 1 1361527 1361528 1 TMEM88B 1 1 1361635 1361636 1 TMEM88B 1 1 1361642 1361726 84 TMEM88B 1 1 1361757 1361761 4 TMEM88B 1 1 1362931 1362956 25 TMEM88B 2 1 1374780 1374838 58 VWA1 3 1 1374841 1374842 1 VWA1 3 1 1374934 1375100 166 VWA1 3 1 1389738 1389747 9 ATAD3C 4 1 1389751 1389816 65 ATAD3C 4 1 1389830 1389849 19 ATAD3C 4 1 1390835 1390837 2 ATAD3C 5 1 1407260 1407331 71 ATAD3B 1 1 1407340 1407474 -

Effects of Chronic Stress on Prefrontal Cortex Transcriptome in Mice Displaying Different Genetic Backgrounds
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Springer - Publisher Connector J Mol Neurosci (2013) 50:33–57 DOI 10.1007/s12031-012-9850-1 Effects of Chronic Stress on Prefrontal Cortex Transcriptome in Mice Displaying Different Genetic Backgrounds Pawel Lisowski & Marek Wieczorek & Joanna Goscik & Grzegorz R. Juszczak & Adrian M. Stankiewicz & Lech Zwierzchowski & Artur H. Swiergiel Received: 14 May 2012 /Accepted: 25 June 2012 /Published online: 27 July 2012 # The Author(s) 2012. This article is published with open access at Springerlink.com Abstract There is increasing evidence that depression signaling pathway (Clic6, Drd1a,andPpp1r1b). LA derives from the impact of environmental pressure on transcriptome affected by CMS was associated with genetically susceptible individuals. We analyzed the genes involved in behavioral response to stimulus effects of chronic mild stress (CMS) on prefrontal cor- (Fcer1g, Rasd2, S100a8, S100a9, Crhr1, Grm5,and tex transcriptome of two strains of mice bred for high Prkcc), immune effector processes (Fcer1g, Mpo,and (HA)and low (LA) swim stress-induced analgesia that Igh-VJ558), diacylglycerol binding (Rasgrp1, Dgke, differ in basal transcriptomic profiles and depression- Dgkg,andPrkcc), and long-term depression (Crhr1, like behaviors. We found that CMS affected 96 and 92 Grm5,andPrkcc) and/or coding elements of dendrites genes in HA and LA mice, respectively. Among genes (Crmp1, Cntnap4,andPrkcc) and myelin proteins with the same expression pattern in both strains after (Gpm6a, Mal,andMog). The results indicate significant CMS, we observed robust upregulation of Ttr gene contribution of genetic background to differences in coding transthyretin involved in amyloidosis, seizures, stress response gene expression in the mouse prefrontal stroke-like episodes, or dementia.