Virucidal Effectiveness Test Using Bovine Viral Diarrhea Virus (BVDV)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Antibiotic-Associated Diarrhea: Candidate Organisms Other Than Clostridium Difficile
The Korean Journal of Internal Medicine : 23:9-15, 2008 Antibiotic-Associated Diarrhea: Candidate Organisms other than Clostridium Difficile Hyun Joo Song, M.D.1, Ki-Nam Shim, M.D.1, Sung-Ae Jung, M.D.1, Hee Jung Choi, M.D.1, Mi Ae Lee, M.D.2, Kum Hei Ryu, M.D.1, Seong-Eun Kim, M.D.1 and Kwon Yoo, M.D.1 Department of Internal Medicine1 and Laboratory Medicine2, Ewha Medical Research Institute, College of Medicine, Ewha Womans University, Seoul, Korea Background/Aims : The direct toxic effects of antibiotics on the intestine can alter digestive functions and cause pathogenic bacterial overgrowth leading to antibiotic-associated diarrhea (AAD). Clostridium difficile (C. difficile) is widely known to be responsible for 10~20% of AAD cases. However, Klebsiella oxytoca, Clostridium perfringens, Staphylococcus aureus, and Candida species might also contribute to AAD. Methods : We prospectively analyzed the organisms in stool and colon tissue cultures with a C. difficile toxin A assay in patients with AAD between May and December 2005. In addition, we performed the C. difficile toxin A assays using an enzyme-linked fluorescent assay technique. Patients were enrolled who had diarrhea with more than three stools per day for at least 2 days after the initiation of antibiotic treatment for up to 6~8 weeks after antibiotic discontinuation. Results : Among 38 patients (mean age 59±18 years, M:F=18:20), the organism isolation rates were 28.9% (11/38) for stool culture, 18.4% (7/38) for colon tissue cultures and 13.2% (5/38) for the C. -

Does Your Patient Have Bile Acid Malabsorption?
NUTRITION ISSUES IN GASTROENTEROLOGY, SERIES #198 NUTRITION ISSUES IN GASTROENTEROLOGY, SERIES #198 Carol Rees Parrish, MS, RDN, Series Editor Does Your Patient Have Bile Acid Malabsorption? John K. DiBaise Bile acid malabsorption is a common but underrecognized cause of chronic watery diarrhea, resulting in an incorrect diagnosis in many patients and interfering and delaying proper treatment. In this review, the synthesis, enterohepatic circulation, and function of bile acids are briefly reviewed followed by a discussion of bile acid malabsorption. Diagnostic and treatment options are also provided. INTRODUCTION n 1967, diarrhea caused by bile acids was We will first describe bile acid synthesis and first recognized and described as cholerhetic enterohepatic circulation, followed by a discussion (‘promoting bile secretion by the liver’) of disorders causing bile acid malabsorption I 1 enteropathy. Despite more than 50 years since (BAM) including their diagnosis and treatment. the initial report, bile acid diarrhea remains an underrecognized and underappreciated cause of Bile Acid Synthesis chronic diarrhea. One report found that only 6% Bile acids are produced in the liver as end products of of British gastroenterologists investigate for bile cholesterol metabolism. Bile acid synthesis occurs acid malabsorption (BAM) as part of the first-line by two pathways: the classical (neutral) pathway testing in patients with chronic diarrhea, while 61% via microsomal cholesterol 7α-hydroxylase consider the diagnosis only in selected patients (CYP7A1), or the alternative (acidic) pathway via or not at all.2 As a consequence, many patients mitochondrial sterol 27-hydroxylase (CYP27A1). are diagnosed with other causes of diarrhea or The classical pathway, which is responsible for are considered to have irritable bowel syndrome 90-95% of bile acid synthesis in humans, begins (IBS) or functional diarrhea by exclusion, thereby with 7α-hydroxylation of cholesterol catalyzed interfering with and delaying proper treatment. -
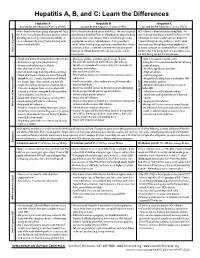
Hepatitis A, B, and C: Learn the Differences
Hepatitis A, B, and C: Learn the Differences Hepatitis A Hepatitis B Hepatitis C caused by the hepatitis A virus (HAV) caused by the hepatitis B virus (HBV) caused by the hepatitis C virus (HCV) HAV is found in the feces (poop) of people with hepa- HBV is found in blood and certain body fluids. The virus is spread HCV is found in blood and certain body fluids. The titis A and is usually spread by close personal contact when blood or body fluid from an infected person enters the body virus is spread when blood or body fluid from an HCV- (including sex or living in the same household). It of a person who is not immune. HBV is spread through having infected person enters another person’s body. HCV can also be spread by eating food or drinking water unprotected sex with an infected person, sharing needles or is spread through sharing needles or “works” when contaminated with HAV. “works” when shooting drugs, exposure to needlesticks or sharps shooting drugs, through exposure to needlesticks on the job, or from an infected mother to her baby during birth. or sharps on the job, or sometimes from an infected How is it spread? Exposure to infected blood in ANY situation can be a risk for mother to her baby during birth. It is possible to trans- transmission. mit HCV during sex, but it is not common. • People who wish to be protected from HAV infection • All infants, children, and teens ages 0 through 18 years There is no vaccine to prevent HCV. -

Bowel Function Anatomy
BOWEL FUNCTION ANATOMY Most of America gives little thought to bowel control. However, bowel control is actually a complex process involving the coordination of many different muscles and nerves. The bowel is considered to be a part of the digestive or gastrointestinal system. It is designed to help the body absorb nutrients and fluids from the foods we eat and drink. After taking out everything the body needs, the bowel then expels the leftover waste. The beginning of the bowel is the small intestine, sometimes referred to as the small bowel. This is where the useful nutrients are absorbed from what you eat. The small bowel delivers the waste to the colon, or large bowel. The colon is a 5-6 foot long muscular tube that delivers stool to the rectum. As the stool moves through the colon, the fluids are removed and absorbed into the body. The consistency of the stool is dependent upon many things, including how long the stool sits in the colon, how much of the water has been absorbed from the waste, and the amount of fiber and fluids in your diet. Stool consistency can vary from hard lumps to mushy to very loose, watery stool. The best and easiest consistency of stool is soft, like toothpaste; this consistency may be attained by adding fiber to your diet. Fiber helps move waste through the colon because it is indigestible by the human body. In other words, fiber adds ‘bulk’ to the stool. It is important to eat a diet high in fiber, however, most Americans lack fiber in their diet. -

Viral Gastroenteritis Backgrounder
Viral Gastroenteritis Backgrounder Viral Gastroenteritis What is Viral Gastroenteritis? Viral gastroenteritis is a stomach illness (including diarrhea and vomiting) in people that is caused by a virus. It is commonly found throughout North America and Europe, and though it can occur year-round, this illness is most often reported in winter. These viruses can also be easily spread in situations of communal living. Viruses are very different from bacteria and parasites. Viruses are much smaller, are not affected by treatment with antibiotics. What are the symptoms of viral gastroenteritis illness? The symptoms of gastroenteritis illness usually include nausea, vomiting, diarrhea, and some stomach cramping. Sometimes people also have a low-grade fever, chills, headache, muscle aches, and a general sense of tiredness. The illness often begins suddenly, and the infected person may feel very sick. The illness is usually brief, with symptoms usually lasting only about 1 or 2 days. In general, children experience more vomiting than adults. Most people with this type of illness have both vomiting and diarrhea. How serious is viral gastroenteritis? Though this type of illness is usually not serious, some people may feel very sick and vomit or have watery diarrhea many times a day. Most people get better within 1 or 2 days, and have no long-term health effects related to their illness; however, if the ill person is unable to drink enough liquids to replace the liquids they lost because of vomiting and diarrhea, they can become dehydrated and may need special medical attention. This problem with dehydration is usually only seen among the very young, the elderly, and persons with weakened immune systems. -
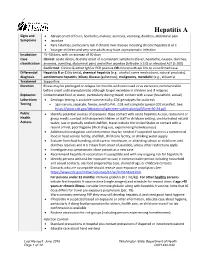
Hepatitis a Reporting Guideline
Hepatitis A Signs and • Abrupt onset of fever, headache, malaise, anorexia, vomiting, diarrhea, abdominal pain Symptoms • Jaundice • Rare fatalities, particularly risk if chronic liver disease including chronic hepatitis B or C • Younger children and very rare adults may have asymptomatic infection Incubation 15–50 days, with an average of 30 days Case Clinical: acute illness, discrete onset of a consistent symptoms (fever, headache, nausea, diarrhea, classification anorexia, vomiting, abdominal pain) and either jaundice (bilirubin ≥ 3.0) or elevated ALT (≥ 200) Confirmed: Clinical & either IgM or PCR positive OR clinical with epi link to a confirmed case Differential Hepatitis B or C (do tests), chemical hepatitis (e.g., alcohol, some medications, natural products), diagnosis autoimmune hepatitis, biliary disease (gallstones), malignancy, metabolic (e.g., Wilson’s) Treatment Supportive Duration Illness may be prolonged or relapse for months with continued virus excretion; communicable before onset until asymptomatic although longer excretion in children and if relapses Exposures Contaminated food or water, particularly during travel; contact with a case (household, sexual) Laboratory • Serologic testing is available commercially; CDC genotypes for outbreak Testing • Spin serum, separate, freeze, send to PHL. CDE will complete special CDC manifest. See: https://www.cdc.gov/laboratory/specimen-submission/pdf/form-50-34.pdf. Public • Identify potential sources of exposure: close contact with acute hepatitis A case, restaurant or Health -

GASTROINTESTINAL COMPLAINT Nausea, Vomiting, Or Diarrhea (For Abdominal Pain – Refer to SO-501) I
DESCHUTES COUNTY ADULT JAIL SO-559 L. Shane Nelson, Sheriff Standing Order Facility Provider: October 17, 2018 STANDING ORDER GASTROINTESTINAL COMPLAINT Nausea, Vomiting, or Diarrhea (for Abdominal Pain – refer to SO-501) I. ASSESSMENT a. History i. Onset and duration ii. Frequency of vomiting, nausea, or diarrhea iii. Blood in stool or black stools? Blood in emesis or coffee-ground appearance? If yes, refer to SO-510 iv. Medications taken – do they help? v. Do they have abdominal pain? If yes, refer to SO-501 Abdominal Pain. vi. Do they have other symptoms – dysuria, urinary frequency, urinary urgency, urinary incontinence, vaginal/penile discharge, hematuria, fever, chills, flank pain, abdominal/pelvic pain in females or testicular pain in males, vaginal or penile lesions/sores? (if yes to any of the above – refer to Dysuria SO-522) vii. LMP in female inmates – if unknown, obtain HCG viii. History of substance abuse? Are they withdrawing? Refer to appropriate SO based on substance history and withdrawal concerns. ix. History of IBS or other known medical causes of chronic diarrhea, nausea, or vomiting? Have prescriptions been used for this in the past? x. History of abdominal surgeries? xi. Recent exposure to others with same symptoms? b. Exam i. Obtain Vital signs, including temperature ii. If complaints of dizziness or lightheadedness with standing, obtain orthostatic VS. iii. Is there jaundice present? iv. Are there signs of dehydration – tachycardia, tachypnea, lethargy, changes in mental status, dry mucous membranes, pale skin color, decreased skin turgor? v. Are you concerned for an Acute Gastroenteritis? Supersedes: March 20, 2018 Review Date: October 2020 Total Pages: 3 1 SO-559 October 17, 2018 Symptoms Exam Viruses cause 75-90% of acute gastroenteritis here in the US. -

Acute Diarrhea in Adults WENDY BARR, MD, MPH, MSCE, and ANDREW SMITH, MD Lawrence Family Medicine Residency, Lawrence, Massachusetts
Acute Diarrhea in Adults WENDY BARR, MD, MPH, MSCE, and ANDREW SMITH, MD Lawrence Family Medicine Residency, Lawrence, Massachusetts Acute diarrhea in adults is a common problem encountered by family physicians. The most common etiology is viral gastroenteritis, a self-limited disease. Increases in travel, comorbidities, and foodborne illness lead to more bacteria- related cases of acute diarrhea. A history and physical examination evaluating for risk factors and signs of inflammatory diarrhea and/or severe dehydration can direct any needed testing and treatment. Most patients do not require labora- tory workup, and routine stool cultures are not recommended. Treatment focuses on preventing and treating dehydra- tion. Diagnostic investigation should be reserved for patients with severe dehydration or illness, persistent fever, bloody stool, or immunosuppression, and for cases of suspected nosocomial infection or outbreak. Oral rehydration therapy with early refeeding is the preferred treatment for dehydration. Antimotility agents should be avoided in patients with bloody diarrhea, but loperamide/simethicone may improve symptoms in patients with watery diarrhea. Probiotic use may shorten the duration of illness. When used appropriately, antibiotics are effective in the treatment of shigellosis, campylobacteriosis, Clostridium difficile,traveler’s diarrhea, and protozoal infections. Prevention of acute diarrhea is promoted through adequate hand washing, safe food preparation, access to clean water, and vaccinations. (Am Fam Physician. 2014;89(3):180-189. Copyright © 2014 American Academy of Family Physicians.) CME This clinical content cute diarrhea is defined as stool with compares noninflammatory and inflamma- conforms to AAFP criteria increased water content, volume, or tory acute infectious diarrhea.7,8 for continuing medical education (CME). -

Pneumatosis Intestinalis in Solid Organ Transplant Recipients
1997 Review Article Pneumatosis intestinalis in solid organ transplant recipients Vincent Gemma1, Daniel Mistrot1, David Row1, Ronald A. Gagliano1, Ross M. Bremner2, Rajat Walia2, Atul C. Mehta3, Tanmay S. Panchabhai2 1Department of Surgery, 2Norton Thoracic Institute, St. Joseph’s Hospital and Medical Center, Phoenix, AZ, USA; 3Department of Pulmonary Medicine, Respiratory Institute, Cleveland Clinic, Cleveland, OH, USA Contributions: (I) Conception and design: D Row, RA Gagliano, TS Panchabhai; (II) Administrative support: RM Bremner, R Walia; (III) Provision of study materials or patients: V Gemma, D Mistrot, TS Panchabhai; (IV) Collection and assembly of data: V Gemma, D Mistrot, TS Panchabhai; (V) Data analysis and interpretation: RA Gagliano, RM Bremner, R Walia, AC Mehta, TS Panchabhai; (VI) Manuscript writing: All authors; (VII) Final approval of manuscript: All authors. Correspondence to: Tanmay S. Panchabhai, MD, FCCP. Associate Director, Pulmonary Fibrosis Center/Co-Director, Lung Cancer Screening Program, Norton Thoracic Institute, St. Joseph’s Hospital and Medical Center, Phoenix, AZ, USA; Associate Professor of Medicine, Creighton University School of Medicine, Omaha, NE, USA. Email: [email protected]. Abstract: Pneumatosis intestinalis (PI) is an uncommon medical condition in which gas pockets form in the walls of the gastrointestinal tract. The mechanism by which this occurs is poorly understood; however, it is often seen as a sign of serious bowel ischemia, which is a surgical emergency. Since the early days of solid organ transplantation, PI has been described in recipients of kidney, liver, heart, and lung transplant. Despite the dangerous connotations often associated with PI, case reports dating as far back as the 1970s show that PI can be benign in solid organ transplant recipients. -

Gastroenteritis Fact Sheet
GASTROENTERITIS FACT SHEET What is Gastroenteritis? Gastroenteritis is an inflammation of the stomach and intestine. There are many causes of Gastroenteritis - some are more serious than others. Viruses, such as Norovirus, are the most common cause of Gastroenteritis and result in vomiting and/or diarrhea. This is often called “stomach flu” but it is not related to the influenza virus. What are the symptoms of viral Gastroenteritis? Most people will get sick between 1 to 2 days after being exposed to the virus. The infection comes on quickly with symptoms of: • Vomiting • Watery diarrhea without blood • Abdominal cramps • Headache • Low-grade fever These symptoms can last for 1 to 10 days depending on the virus. Do I need treatment? Most people get better without any problems. Young children, elders and people with health problems may be at risk for dehydration. • People become dehydrated when they do not drink enough liquids to replace the fluids they are losing from vomiting and having diarrhea. Adults with dehydration will feel thirsty, go pee less often and feel dizzy when standing up. Avoid drinking fluids with caffeine (e.g., coffee and pop). Avoid drinking alcohol. These beverages can make your dehydration worse. Reasons to contact your health care provider: • If the diarrhea is bloody • If there is high fever (over 38 degrees Celsuis) • If you think you or someone you are caring for is more seriously dehydrated, contact your health care provider or go to the emergency room. How do I get viral Gastroenteritis? Viruses that cause Gastroenteritis are found in the stool and sometimes in the vomit of someone who is sick. -

Traveler's Diarrhea
Traveler’s Diarrhea JOHNNIE YATES, M.D., CIWEC Clinic Travel Medicine Center, Kathmandu, Nepal Acute diarrhea affects millions of persons who travel to developing countries each year. Food and water contaminated with fecal matter are the main sources of infection. Bacteria such as enterotoxigenic Escherichia coli, enteroaggregative E. coli, Campylobacter, Salmonella, and Shigella are common causes of traveler’s diarrhea. Parasites and viruses are less common etiologies. Travel destination is the most significant risk factor for traveler’s diarrhea. The efficacy of pretravel counseling and dietary precautions in reducing the incidence of diarrhea is unproven. Empiric treatment of traveler’s diarrhea with antibiotics and loperamide is effective and often limits symptoms to one day. Rifaximin, a recently approved antibiotic, can be used for the treatment of traveler’s diarrhea in regions where noninvasive E. coli is the predominant pathogen. In areas where invasive organisms such as Campylobacter and Shigella are common, fluoroquinolones remain the drug of choice. Azithromycin is recommended in areas with qui- nolone-resistant Campylobacter and for the treatment of children and pregnant women. (Am Fam Physician 2005;71:2095-100, 2107-8. Copyright© 2005 American Academy of Family Physicians.) ILLUSTRATION BY SCOTT BODELL ▲ Patient Information: cute diarrhea is the most com- mised and those with lowered gastric acidity A handout on traveler’s mon illness among travelers. Up (e.g., patients taking histamine H block- diarrhea, written by the 2 author of this article, is to 55 percent of persons who ers or proton pump inhibitors) are more provided on page 2107. travel from developed countries susceptible to traveler’s diarrhea. -

Antibiotic-Associated Diarrhea Patient Information
ANTIBIOTIC-ASSOCIATED DIARRHEA PATIENT INFORMATION If you have received antibiotics while in hospital, or have been prescribed antibiotics that you are to take following discharge from hospital, please review this information sheet on antibiotic-associated diarrhea. If you have any questions, ask your nurse, doctor, or pharmacist. ____________________________________________________________________________________________ Antibiotics can cause diarrhea in up to one third of people who take them. Most often, the diarrhea is mild. Sometimes, a more serious type of diarrhea associated with taking antibiotics is caused by the Clostridium difficile bacterium. Why can diarrhea occur with antibiotics? Bacteria are normally present in your bowel. Diarrhea can occur because antibiotics kill some of the bacteria that usually live in your bowel. This upsets the normal balance. Sometimes harmful bacteria such as Clostridium difficile, if present in your bowel, can overgrow leading to diarrhea and other symptoms. The risk of Clostridium difficile is higher if you have been in the hospital. What are the symptoms? Diarrhea from antibiotics is usually mild, consisting of loose and/or frequent bowel movements. Symptoms of Clostridium difficile diarrhea may be more severe and may include: • Watery diarrhea that may contain mucus and/or blood • Abdominal pain or tenderness • Loss of appetite • Nausea • Fever • What should you do if you get diarrhea? If you are taking an antibiotic and have mild diarrhea that is not bothersome and you are able to eat and drink without difficulty, continue to take the antibiotic as prescribed. The diarrhea should go away after the antibiotic is finished. CALLYOUR DOCTOR IF you have any of the following symptoms: • Diarrhea which is bothersome or severe, or which is bloody • Abdominal pain • Fever • Diarrhea which continues after the antibiotic is finished • Diarrhea which starts after you have finished taking the antibiotic(s).