14. Galc Activity 94
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Outlaw: Wilderness and Exile in Old and Middle
THE ‘BESTLI’ OUTLAW: WILDERNESS AND EXILE IN OLD AND MIDDLE ENGLISH LITERATURE A Dissertation Presented to the Faculty of the Graduate School of Cornell University In Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy by Sarah Michelle Haughey August 2011 © 2011 Sarah Michelle Haughey THE ‘BESTLI’ OUTLAW: WILDERNESS AND EXILE IN OLD AND MIDDLE ENGLISH LITERATURE Sarah Michelle Haughey, Ph. D. Cornell University 2011 This dissertation, The ‘Bestli’ Outlaw: Wilderness and Exile in Old and Middle English Literature explores the reasons for the survival of the beast-like outlaw, a transgressive figure who highlights tensions in normative definitions of human and natural, which came to represent both the fears and the desires of a people in a state of constant negotiation with the land they inhabited. Although the outlaw’s shelter in the wilderness changed dramatically from the dense and menacing forests of Anglo-Saxon England to the bright, known, and mapped greenwood of the late outlaw romances and ballads, the outlaw remained strongly animalistic, other, and liminal, in strong contrast to premodern notions of what it meant to be human and civilized. I argue that outlaw narratives become particularly popular and poignant at moments of national political and ecological crisis—as they did during the Viking attacks of the Anglo-Saxon period, the epoch of intense natural change following the Norman Conquest, and the beginning of the market revolution at the end of the Middle Ages. Figures like the Anglo-Saxon resistance fighter Hereward, the exiled Marcher lord Fulk Fitz Waryn, and the brutal yet courtly Gamelyn and Robin Hood, represent a lost England imagined as pristine and forested. -

Fraternization: Time for a Rational Department of Defense Standard
Volume 135 Winter 1992 MILITARY LAW REVIEW ARTICLES IS THE DOCTRISE OF EQIXABLE TOLLISG APPLICABLE TO THE LIYITATIOSSPERIODS IS THE FEDERAL TORT CLAIMS ACT? . , . , . , . , . , , . , . , , , . , , . , . , . , , . , . , . , , , , . , , , , , . , . Richard Parker CRIMISALL IABILITY CSDERTHE USIFORMC ODE OF MILITARY JUSTICE FOR SEXL.4L RELATIOSS DCRISGP SYCHOTHERAPY . , . , . , . , . , . , . , , . , . , . , . , . , . , . , . , , . Captain Jody M. Prescott Dr. Matthew G. Snow FRATERSIZATION:TIME FOR A RATIOS DEPARTMENT OF UEFESSE STANDARD Major David S. Jonas FREEDOM OF ~AVIG.4TION.4SD THE BLACK SEA BCMPINGISCIDENT: How "INSOCEST"M UST ISNOCENTPASSAGE BE Lieutenant Commander John W. Rolph DETERMISING CLEASUPSTASDARDS FOR HAZARDOLS WASTE SITES , . , , . , . , . , . , . , . , . , . , . , . , . , . , Major William D. Turkula BOOK REVIEWS Charlottesville, Virginia Pamphlet HEADQUARTERS DEPARTMENT OF THE ARMY NO. 27-100-135 Washington, D.C., Winter 1992 MILITARY LAW REVIEW- VOLUME 135 The Military Law Review has been published quarterly at The Judge Advocate General’s School, US. Army, Charlottes- ville, Virginia, since 1958. The Review provides a forum for those interested in military law to share the products of their experiences and research and is designed for use by military attorneys in connection with their official duties. Writings of- fered for publication should be of direct concern and import in this area of scholarship, and preference will be given to writ- ings that have lasting value as reference materials for the mili- tary lawyer. The Review encourages frank discussion of rele- vant legislative, administrative, and judicial developments. EDITORIAL STAFF MAJOR DANIEL P. SHAVER, Editor MS. EVA F. SKINNER, Editorial Assistant SUBSCRIPTIONS: Private subscriptions may be purchased from the Superintendent of Documents, United States Govern- ment Printing Office, Washington, D.C. 20402. Publication ex- change subscriptions are available to law schools and other organizations that publish legal periodicals. -

List of Section 13F Securities
List of Section 13F Securities 1st Quarter FY 2004 Copyright (c) 2004 American Bankers Association. CUSIP Numbers and descriptions are used with permission by Standard & Poors CUSIP Service Bureau, a division of The McGraw-Hill Companies, Inc. All rights reserved. No redistribution without permission from Standard & Poors CUSIP Service Bureau. Standard & Poors CUSIP Service Bureau does not guarantee the accuracy or completeness of the CUSIP Numbers and standard descriptions included herein and neither the American Bankers Association nor Standard & Poor's CUSIP Service Bureau shall be responsible for any errors, omissions or damages arising out of the use of such information. U.S. Securities and Exchange Commission OFFICIAL LIST OF SECTION 13(f) SECURITIES USER INFORMATION SHEET General This list of “Section 13(f) securities” as defined by Rule 13f-1(c) [17 CFR 240.13f-1(c)] is made available to the public pursuant to Section13 (f) (3) of the Securities Exchange Act of 1934 [15 USC 78m(f) (3)]. It is made available for use in the preparation of reports filed with the Securities and Exhange Commission pursuant to Rule 13f-1 [17 CFR 240.13f-1] under Section 13(f) of the Securities Exchange Act of 1934. An updated list is published on a quarterly basis. This list is current as of March 15, 2004, and may be relied on by institutional investment managers filing Form 13F reports for the calendar quarter ending March 31, 2004. Institutional investment managers should report holdings--number of shares and fair market value--as of the last day of the calendar quarter as required by Section 13(f)(1) and Rule 13f-1 thereunder. -

Scriptedpifc-01 Banijay Aprmay20.Indd 2 10/03/2020 16:54 Banijay Rights Presents… Bäckström the Hunt for a Killer We Got This Thin Ice
Insight on screen TBIvision.com | April/May 2020 Television e Interview Virtual thinking The Crown's Andy Online rights Business Harries on what's companies eye next for drama digital disruption TBI International Page 10 Page 12 pOFC TBI AprMay20.indd 1 20/03/2020 20:25 Banijay Rights presents… Bäckström The Hunt For A Killer We Got This Thin Ice Crime drama series based on the books by Leif GW Persson Based on a true story, a team of police officers set out to solve a How hard can it be to solve the world’s Suspense thriller dramatising the burning issues of following the rebellious murder detective Evert Bäckström. sadistic murder case that had remained unsolved for 16 years. most infamous unsolved murder case? climate change, geo-politics and Arctic exploitation. Bang The Gulf GR5: Into The Wilderness Rebecka Martinsson When a young woman vanishes without a trace In a brand new second season, a serial killer targets Set on New Zealand’s Waiheke Island, Detective Jess Savage hiking the famous GR5 trail, her friends set out to Return of the riveting crime thriller based on a group of men connected to a historic sexual assault. investigates cases while battling her own inner demons. solve the mystery of her disappearance. the best-selling novels by Asa Larsson. banijayrights.com ScriptedpIFC-01 Banijay AprMay20.indd 2 10/03/2020 16:54 Banijay Rights presents… Bäckström The Hunt For A Killer We Got This Thin Ice Crime drama series based on the books by Leif GW Persson Based on a true story, a team of police officers set out to solve a How hard can it be to solve the world’s Suspense thriller dramatising the burning issues of following the rebellious murder detective Evert Bäckström. -

Iky Jn M Pato Pont Mísb the Biú Stft Parad This Morning at JO O'clock
Today Iky Jn M Pato Pont Mísb The Biú Stft Parad This Morning at JO o'Clock rRrwTrjaelt, iaVmr 4 . ... YEAR. SPAMSH; SECTION PAbjO, - 4 PAGES EL; TEXiAfl.- WEDhffi3&AYt OCTOBER 28, 1914. SECTION 12 PAGES. PRICE 5 CENTS ' . "' -T ,. M E!3H DAY'S SUMMARY PLOT; WMiAirs ifsws mm AGAÍNS HUI T8 W)rVIWWW. OF EUROPEAN WAR IF VILLA WILL ' te tfci süte UfVeatfMi AH the halloas engaged m thé migMy straggle m Helglnm and Hte north or Ftaare are silent on the their dear-- aeWrl hsfprnlags m that d LIFE rrjr.4 4he TeiuM , go flf VOLA United BiMMtn afj Iba tone, tar as Is known, I Centederaey. The member at ar there bss been Rule progress en III bavsebsM, .new fertjr-iw- hi swaaber, rtthervelder but-- freat iba aeeowte AI hi eensideraMen, ef tbehr atea, are H KELLY thst have tillered through treat neMent beetth;. The tbembiWmea-sates- i this stern engagement, M tent ta the besae staring the wbfeR has been going en Incessantly ee" by the Chapters' Texas far several davit, may be character- ised aa the fiercest of the whole war. OUT CARRANZA Theaundt upon thousands of Ger- . .The eks jeai .te )fce.heeie jby the man rttatorcements have been added ebaptera, m respenae t ny call nade te the great masses of troops which 'ta the flan' Antéale eeaveatlen year have been endeavoring te force their age, have giren, mere, pteaanre to the way la the northern porta el rraaee. WILL LIKEWISE eMladlea than eny'resaeabraaea ever it ts said that this eeesetess push- v i 4. -

EDISON-RS-2012-ENG Letter.Pdf
Sustainability Report Sustainability Report 2012 2012 Edison Spa Foro Buonaparte, 31 20121 Milan tel. +39 02 6222.1 www.edison.it Contents 4 Growth despite the crisis 6 Edison Sustainable Development Policy 47 People as a resource 7 The challenges, the goals 48 Empower the human capital 50 Choosing to improve 54 Health and safety 13 We at Edison. 56 Industrial relations 14 Energy and responsibility 57 Personnel involvement 16 Actors on today’s stage 18 Activities and Projects in the Hydrocarbon Sector 22 Sustainability and Governance 59 The market is our benchmark 27 Stakeholders: our point of reference 60 Edison’s Product Offers for the Market 29 The wealth we create 64 The quality of customer service 67 We seek for comparison 31 Environment means responsibility 32 Our commitments to the environment 69 Respecting the community 34 Mitigating significant environmental impacts 70 Local community relations 39 A systematic approach to biodiversity 82 Shareholders and financers 84 Suppliers 86 Institutions 89 Note on methodology 90 Performance indicators 104 GRI Index 108 Report of the Independent Auditors 110 Edison on line Sustainability Report 2012 Sustainability Report Edison 2012 Edison in Italy... HEADQUARTERS OPERATING COMPANY KHR PLANT (Edison 20%) PRATI DI VIZZE THERMOELECTRIC POWER PLANT EL.IT.E CURON BRUNICO HYDROELECTRIC POWER PLANT GLORENZA MARLENGO WIND FARM LASA PONTE GARDENA SONICO CASTELBELLO PREMESA PHOTOVOLTAIC SYSTEM PIEVE CAMPO BELVISO TAIO VERGONTE CEDEGOLO VENINA R&D CENTER ALBANO MEZZOCORONA VAL MEDUNA (5 plants) BATTIGGIO ARMISA GANDA POZZOLAGO MERCHANT LINE EL.I.TE. VEDELLO CIVIDATE TORVISCOSA MONZA ZAPPELLO VAL CAFFARO (4 plants) COLLALTO COLOGNO MONZESE PUBLINO Selvazzano MARGHERA COMPRESSOR STATION SESTO S.G. -

Omnibus Codes
Medical Coverage Policy Effective Date ............................................. 7/15/2021 Next Review Date ......................................11/15/2021 Coverage Policy Number .................................. 0504 Omnibus Codes Table of Contents Related Coverage Resources Overview .............................................................. 1 Category III Current Procedural Terminology (CPT®) Coverage Policy ................................................... 1 codes General Background ............................................ 7 Deep Brain and Motor Cortex and Responsive Services without Food and Drug Cortical Stimulation Administration (FDA) Approval ....................... 7 Electrodiagnostic Testing (EMG/NCV) Cardiovascular ................................................ 7 Serological Testing for Inflammatory Bowel Disease Gastroenterology .......................................... 36 Neurology ...................................................... 61 Obstetrics/Gynecology .................................. 67 Urology .......................................................... 69 Ophthalmology .............................................. 72 Oncology ....................................................... 80 Otolaryngology .............................................. 86 Other ............................................................. 89 INSTRUCTIONS FOR USE The following Coverage Policy applies to health benefit plans administered by Cigna Companies. Certain Cigna Companies and/or lines of business only provide -

Serie Categoría Temporadas
Serie Categoría Temporadas Industry Series Drama 1 WandaVision Series Ciencia Ficcion 1 The Flight Attendant Series Drama 1 Eliminatorias Qatar 2022 Series Accion 1 The Stand Series Ciencia Ficcion 1 Bridgerton Series Drama 1 The Wilds Series Drama 1 Love Life Series Comedia 1 Truth Seekers Series Comedia 1 Equinox Series Misterio 1 El Cid Series Accion 1 Dickinson Series Comedia 1 Catalina la Grande Series Drama 1 La Revolución Series Drama 1 Selena: La serie Series Drama 1 Superstore Series Comedia 4 Antidisturbios Series Drama 1 The Crown Series Drama 4 Patria Series Drama 1 Monsterland Series Terror 1 Los favoritos de Midas Series Misterio 1 De brutas, nada Series Comedia 1 The Liberator Series Accion 1 Dash y Lily Series Drama 1 Dignidad Series Drama 1 Miénteme Series Drama 3 Wireless Series Misterio 1 Gambito de dama Series Drama 1 High School Musical: El Musical: La Serie Series Comedia 1 The Runaways Series Aventura 3 Alien TV Series Animadas 1 Amor mío Series Novelas 1 Gigantes de la industria Series Documentales 1 Nuevo Rico Nuevo Pobre Series Novelas 1 The Walking Dead: World Beyond Series Ciencia Ficcion 1 Raised by Wolves Series Ciencia Ficcion 1 Digimon Series Animadas 5 Pennyworth Series Drama 1 One Piece Series Animadas 1 Hightown Series Drama 1 Bates Motel Series Misterio 5 Alguien tiene que morir Series Drama 1 Warrior Series Accion 1 La maldición de Bly Manor Series Misterio 1 Normal People Series Drama 1 Mañana es para siempre Series Novelas 1 En Nombre Del Amor Series Novelas 0 Escalera al cielo Series Novelas 1 Hacia -

Open Tamul Dissertation.Pdf
The Pennsylvania State University The Graduate School College of Communications IMMIGRATION NEWS FRAMES: CONSTRUCTIONS OF IMMIGRATION AND THE PASSAGE OF ARIZONA SB 1070 A Dissertation in Mass Communications by Daniel J. Tamul © 2011 Daniel J. Tamul Submitted in Partial Fulfillment of the requirements for the Degree of Doctor of Philosophy August 2011 ii The dissertation of Daniel J. Tamul was reviewed and approved* by the following: Dennis K. Davis Professor of Communications Dissertation Adviser Chair of Committee Matthew McAllister Professor of Communications Colleen Connolly-Ahern Associate Professor of Communications Gretchen Casper Associate Professor of Political Science Marie Hardin Associate Dean for Undergraduate and Graduate Education *Signatures are on file in the Graduate School. iii Abstract This study examined the news frames surrounding coverage of immigration and immigrants, specifically undocumented immigrants, in the time surrounding the passage of a particularly controversial immigration bill, Arizona SB 1070. An inductive framing analysis and longitudinal discourse analysis were conducted to develop a comprehensive understanding of the frames used in news accounts within the state of Arizona and, more broadly, in national news coverage. Generally speaking, immigration and immigrants are socially constructed in news accounts as a detrimental influence on society at large. These frames serve to naturalize and justify the passage of policies that may lead to discriminatory practices. Practical and theoretical implications -

Information to Users
INFORMATION TO USERS This manuscript has been reproduced from the microfihn master. UMI films the text directly from the original or copy submitted. Thus, some thesis and dissertation copies are in typewriter face, while others may be from any type o f computer printer. The quality of this reproduction is dependent upon the quality of the copy submitted. Broken or indistinct print, colored or poor quality illustrations and photographs, print bleedthrough, substandard margins, and improper alignment can adversely affect reproduction. In the unlikely event that the author did not send UMI a complete manuscript and there are missing pages, these will be noted. Also, if unauthorized copyright material had to be removed, a note will indicate the deletion. Oversize materials (e.g., maps, drawings, charts) are reproduced by sectioning the original, beginning at the upper left-hand comer and continuing from left to right in equal sections with small overlaps. Each original is also photographed in one exposure and is included in reduced form at the back o f the book. Photographs included in the original manuscript have been reproduced xerographically in this copy. Higher quality 6” x 9” black and white photographic prints are available for any photographs or illustrations appearing in this copy for an additional charge. Contact UMI directly to order. UMI A Bell A Knvell Infoimition Company 300 North Zeeb Road, Ann Arbor MI 48106-1346 USA 313/761-4700 800/5214)600 THE SYNCHRONY AND DIACHRONY OF ENGLISH IMPERSONAL VERBS: A STUDY IN SYNTACTIC AND LEXICAL CHANGE DISSERTATION Presented in Partial Fulfillment of the Requirement for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By Hyeree Kim, B.A., M.A. -

View (E16460) Karen Fortuna, John Naslund, Jessica Lacroix, Cynthia Bianco, Jessica Brooks, Yaara Zisman-Ilani, Anjana Muralidharan, Patricia Deegan
JMIR Mental Health Internet interventions, technologies and digital innovations for mental health and behaviour change Volume 7 (2020), Issue 4 ISSN: 2368-7959 Editor in Chief: John Torous, MD Contents Editorial Advancing E-Mental Health in Canada: Report From a Multistakeholder Meeting (e19360) Gillian Strudwick, Danielle Impey, John Torous, Reinhard Krausz, David Wiljer. 3 Original Papers A Web- and Mobile-Based Intervention for Comorbid, Recurrent Depression in Patients With Chronic Back Pain on Sick Leave (Get.Back): Pilot Randomized Controlled Trial on Feasibility, User Satisfaction, and Effectiveness (e16398) Sandra Schlicker, Harald Baumeister, Claudia Buntrock, Lasse Sander, Sarah Paganini, Jiaxi Lin, Matthias Berking, Dirk Lehr, David Ebert. 6 Temporal Associations of Daily Changes in Sleep and Depression Core Symptoms in Patients Suffering From Major Depressive Disorder: Idiographic Time-Series Analysis (e17071) Noah Lorenz, Christian Sander, Galina Ivanova, Ulrich Hegerl. 27 A Web-Based Adaptation of the Quality of Life in Bipolar Disorder Questionnaire: Psychometric Evaluation Study (e17497) Emma Morton, Sharon Hou, Oonagh Fogarty, Greg Murray, Steven Barnes, Colin Depp, CREST.BD, Erin Michalak. 38 Digital Peer Support Mental Health Interventions for People With a Lived Experience of a Serious Mental Illness: Systematic Review (e16460) Karen Fortuna, John Naslund, Jessica LaCroix, Cynthia Bianco, Jessica Brooks, Yaara Zisman-Ilani, Anjana Muralidharan, Patricia Deegan. 65 The Performance of Emotion Classifiers for Children -
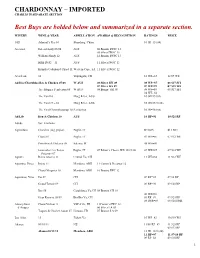
Chardonnay – Imported Chablis in Separate Section
CHARDONNAY – IMPORTED CHABLIS IN SEPARATE SECTION Best Buys are bolded below and summarized in a separate section. WINERY WINE & YEAR APPELLATION AWARDS & RECOGNITION RATINGS PRICE 1421 Admiral’s Res 10 Shandong, China 10 JR=15 (84) Accolade Eileen Hardy 05/06 AUS 06 Bronze IWSC 13 05 Silver IWSC 13 William Hardy 12 AUS 12 Bronze IWSC 13 HRB D652 – 11 AUS 11 Silver IWSC 13 Kumala Colunbard-Chard 11 Western Cape, SA 11 Silver IWSC 12 Acordeon 12 Tupungato, CH 12 WE=83 12 $9 WE Ad Hoc/CherubinoHen & Chicken 07/08 W AUS 08 Silver SFI 09 08 WS=87 08 $17 SFI 07 Silver DA 09 07 WS=89 07 $18 WS Tree Hugger Pemberton 08 W AUS 08 Bronze SFI 09 08 WS=88 08 $17 SFI 08 WE=82 The Yard 10 Marg River, AUS 10 JR=15 (84) The Yard 24 – 10 Marg River, AUS 10 JR=15.5 (86) The Yard Channybeaurup 10 Pemberton 10 JR=16 (88) Ad Lib Hen & Chicken 10 AUS 10 RP=91 10 $21 RP Adobe See: Emiliana Agrinatura Chard nv (org grapes) Puglia, IT BTI=85 $13 BTI Chard 07 Puglia, IT 07 WE=86 07 $12 WE Primitivo del Salento 08 Salento, IT 08 WE=86 Giancarlo Ceci Pozzo Puglia, IT 07 Editor’s Choice WE 10/21/08 07 WE=87 07 $14 WE Sorgente 07 Aguirre Reina Ana res 11 Central Va, CH 11 WE=84 11 $12 WE Agostino, Finca Inicio 11 Mendoza, ARH 11 Comm’d Decanter 12 Chard-Viognier 10 Mendoza, ARG 10 Bronze IWC 12 Agustinos, Vina Est 09 CH 09 RP=85 09 $8 RP Grand Terroir 09 CH 09 RP=90 09 $20 RP Res 08 Casablanca Va,CH 08 Bronze CH 10 06 WE=86 Gran Reserva 08/09 Bio Bio Va, CH 09 RP=89 09 $25 RP 08 IWR=89 08 $20 IWR AimerySieur Chard Vivhon 11 VdP d’Oc, FR 11Comm’d IWC 12 d’Arquea 06