Mutational Profile of Malignant Pleural Mesothelioma
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

CV Francesco Patti Name Francesco Patti Born in Acireale (Italy) 12/06/1958 Scholastic Career II Level Education 1976 Degree Me
Name Francesco Patti Born in Acireale (Italy) 12/06/1958 Scholastic career II level education 1976 Degree Medicine and Surgery 1982 maximum cum laude Post Degree Neurology, University of Catania, 1986 Post Degree Phyisiotherapy, University of Parma, 1990 Work activity 1987-1988 Regional fellowship as Junior Neurologist to study “ Descriptive Neuroepidemiology of most frequent neurological diseases in Sicily”, progetto Regionale 55/P, sponsored by WHO. 1987–1989 Assisting Professor of Neuroendocrinology and Neuroimmunology Scuola di Specializzazione in Neurologia, University of Catania 1991–2000 “CollaboratoreTecnico” Chair of Neurorheabilitation, Institute of Neurological Sciences, University of Catania. 2000–2002 “Tecnico Laureato” (Funzionario Tecnico) Department of Neurological Sciences November 2002 – October 2014 “Ricercatore Confermato” (Aggregate Professor) Clinical Researcher Department of Neurological Sciences, University of Catania. November 2014 - up till now Associate Professor of Neurology, Department of Medical and Surgical Sciences and Advanced Tecnologies G.F. Ingrassia, University of Catania. Clinician profile and activities He is responsible of the tertiary centre of multiple sclerosis at the University of Catania (Italy). The centre follows more than 2500 patients suffering from Multiple Sclerosis and few patients suffering from Devic Disease and Devic Spectrum disorder diseases. The centre follows also patients suffering from ALS (currently 100 patients) and other people with different forms of spasticity, offering them with a multidisciplinary approach every kind of Pag. 1 of 3 CV Francesco Patti You created this PDF from an application that is not licensed to print to novaPDF printer (http://www.novapdf.com) assistance. Scientific activity Research Interests ; Preclinical (1981-1988) ; Neurochemistry, Neuroendocrinolgy, Neuropsycopharmacology ; Clinical: (1989-current) ; Neuroepidemiology ; Clinical Immunology ; Quality of Life ; Neurorehabilitation ; Multiple Sclerosis. -
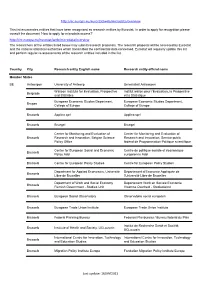
Eurostat: Recognized Research Entity
http://ec.europa.eu/eurostat/web/microdata/overview This list enumerates entities that have been recognised as research entities by Eurostat. In order to apply for recognition please consult the document 'How to apply for microdata access?' http://ec.europa.eu/eurostat/web/microdata/overview The researchers of the entities listed below may submit research proposals. The research proposal will be assessed by Eurostat and the national statistical authorities which transmitted the confidential data concerned. Eurostat will regularly update this list and perform regular re-assessments of the research entities included in the list. Country City Research entity English name Research entity official name Member States BE Antwerpen University of Antwerp Universiteit Antwerpen Walloon Institute for Evaluation, Prospective Institut wallon pour l'Evaluation, la Prospective Belgrade and Statistics et la Statistique European Economic Studies Department, European Economic Studies Department, Bruges College of Europe College of Europe Brussels Applica sprl Applica sprl Brussels Bruegel Bruegel Center for Monitoring and Evaluation of Center for Monitoring and Evaluation of Brussels Research and Innovation, Belgian Science Research and Innovation, Service public Policy Office fédéral de Programmation Politique scientifique Centre for European Social and Economic Centre de politique sociale et économique Brussels Policy Asbl européenne Asbl Brussels Centre for European Policy Studies Centre for European Policy Studies Department for Applied Economics, -

Creating Value in the Entrepreneurial University: Marketization and Merchandising Strategies
administrative sciences Article Creating Value in the Entrepreneurial University: Marketization and Merchandising Strategies Chiara Fantauzzi *, Rocco Frondizi , Nathalie Colasanti and Gloria Fiorani Department of Management and Law, University of Rome Tor Vergata, 00133 Roma, Italy; [email protected] (R.F.); [email protected] (N.C.); gloria.fi[email protected] (G.F.) * Correspondence: [email protected] Received: 9 August 2019; Accepted: 14 October 2019; Published: 18 October 2019 Abstract: Higher education institutions are called to expand their role and responsibilities, by enhancing their entrepreneurial mindset and redefining relationships with stakeholders. In order to cope with these new challenges, they have started to operate in a strategic manner, by performing marketing and merchandising activities. Indeed, in a sector characterized by the presence of competitive funding models, several forms of accountability, and performance indicators, universities have become open systems and have started to operate like enterprises, considering students as customers. Given this premise, the aim of the paper is to individuate marketing and merchandising strategies in higher education and to evaluate their effectiveness in order to foster stakeholders engagement. This is in line with the entrepreneurial university model that represents the starting point of the theoretical study, then a literature review of “marketization” in higher education institutions is presented, showing how this field is not yet completely investigated. Data refer to the Italian context and are analyzed through a qualitative method. Findings suggest that most Italian universities perform merchandising strategies, but currently there is not sufficient information to evaluate their effectiveness in higher education, it was only possible to make hypotheses. -

Folder Ciiscam Solo Inglese
Department of Ecology and Economic Sustainable Development Under the High Patronage of The President of the Republic of Italy ITALIAN OFFICIAL WORLD FOOD DAY CELEBRATIONS 2007 The Right to Food Under the Patronage of The City of Viterbo The Province of Viterbo The Agriculture Commission of the Region of Lazio The Chair of the Council of the Lazio Region The Ministry of Agriculture 1° INTERNATIONAL C.I.I.S.C.A.M.CONFERENCE IINTERNATIONAL INTER-UNIVERSITY CENTRE FOR MEDITERRANEAN FOOD CULTURE STUDIES New Frontiers in the Mediterranean for Food Security Mediterranean Diet and Well Being Food Safety and Quality Biodiversity and Nutrition 4-5 December 2007 Rector Hall, via Santa Maria in Gradi, 4 Viterbo in cooperation with FORUM ON National Institute for Research Nutrition and Consumer MEDITERRANEAN Italian Official Celebrations on Food and Nutrition Protection Division FOOD CULTURES WORLD FOOD DAY 2007 1° INTERNATIONAL CIISCAM CONFERENCE CIISCAM INTERUNIVERSITY INTERNATIONAL CENTRE FOR MEDITERRANEAN FOOD CULTURES STUDIES The CIISCAM - Interuniversity International Centre for OBJECTIVES: Mediterranean Food Cultures Studies - has been esta- - To promote, realize and coordinate researches in the blished on 25 July 2006 by the Sapienza University of field of food science, with particular regards to Rome, the University of Calabria, the University of Gran Mediterranean food cultures; Canaria, the University of Parma and the University of - To foster cooperation among participant universities Tuscia. Its administrative office is at the Sapienza -

Welcome Package for Erasmus and International Students
WELCOME PACKAGE FOR ERASMUS AND INTERNATIONAL STUDENTS University of Parma - Main Building 2 WELCOME TO THE UNIVERSITY OF PARMA We are delighted that you have chosen to study at the University of Parma. We hope that your time here will be challenging, academically rewar- ding and enjoyable. This Welcome Guide is designed to help you manage the process of planning and moving to Parma for your studying experience. It provi- des you with instructions and guidance on visa application, registra- tion procedures, admission requirements for the courses and on the services and facilities at your disposal. Moreover, this booklet includes practical information for your arrival and your stay in Parma, as well as maps and important contact details. We have also added a specific section on Parma, containing handy tips on what to see and do in your free time. We look forward to meeting and welcoming you to the University of Parma. The University of Parma has been awarded the ECTS Label twice in a row, for the period 2009-2013 and 2013-2016. 3 Table of CONTENTS Why choosing the University of Parma 6 APPLYING FOR UNIPR 9 How to apply - Exchange Students 10 Before your arrival - Exchange Students 12 On arrival - Exchange Students 12 At the end of your exchange period 14 How to apply - International Degree-seeking Students 16 Application procedure 20 Visa application 28 Registration to the town council records 32 Residence permit 33 Italian tax code - codice fiscale 35 STUDYING AT UNIPR 36 Academic calendar 37 The University Departments 38 Course catalogue -

Unveiling Role of Sphingosine-1-Phosphate Receptor 2 As a Brake of Epithelial Stem Cell Proliferation and a Tumor Suppressor in Colorectal Cancer
Unveiling role of Sphingosine-1-phosphate receptor 2 as a brake of epithelial stem cell proliferation and a tumor suppressor in colorectal cancer Luciana Petti Humanitas Clinical and Research Center-IRCCS Giulia Rizzo Humanitas University Federica Rubbino Humanitas University Sudharshan Elangovan Humanitas University Piergiuseppe Colombo Humanitas Clinical and Research Center-IRRCS Restelli Silvia Humanitas University Andrea Piontini Humanitas University Vincenzo Arena Policlinico Universitario Agostino Gemelli Michele Carvello Humanitas Clinical and Research Center-IRCCS Barbara Romano Universita degli Studi di Napoli Federico II Dipartimento di Medicina Clinica e Chirurgia Tommaso Cavalleri Humanitas Clinical and Research Center-IRCCS Achille Anselmo Humanitas Clinical and Research Center-IRCCS Federica Ungaro Humanitas University Silvia D’Alessio Humanitas University Antonino Spinelli Humanitas University Sanja Stifter Page 1/28 University of Rijeka Fabio Grizzi Humanitas Clinical and Research Center-IRCCS Alessandro Sgambato Istituto di Ricovero e Cura a Carattere Scientico Centro di Riferimento Oncologico della Basilicata Silvio Danese Humanitas University Luigi Laghi Universita degli Studi di Parma Alberto Malesci Humanitas University STEFANIA VETRANO ( [email protected] ) Humanitas University Research Keywords: colorectal cancer, Lgr5, S1PR2, PTEN, epithelial proliferation Posted Date: October 13th, 2020 DOI: https://doi.org/10.21203/rs.3.rs-56319/v2 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Version of Record: A version of this preprint was published on November 23rd, 2020. See the published version at https://doi.org/10.1186/s13046-020-01740-6. Page 2/28 Abstract Background. Sphingosine-1-phosphate receptor 2 (S1PR2) mediates pleiotropic functions encompassing cell proliferation, survival, and migration, which become collectively de-regulated in cancer. -

Alessandro Tasora
Curriculum Vitae Alessandro Tasora Born 6-3-1971 in Milano. Fiscal code: TSRLSN71C06F205H v. Avesella 26, 40121 Bologna, ITALY [email protected] http://projectchrono.org/tasora/ http://www.chronoengine.info WORK EXPERIENCE 2014- Associate Professor, at the Department of Industrial Engineering, University of Parma, Italy. 2002-2014 Assistant Professor, at the Department of Industrial Engineering, University of Parma, Italy. 1998-2002 Researcher at the Dipartimento di Sistemi di Trasporto e Movimentazione and Dipartimento di Elettrotecnica, at Politecnico di Milano, Italy. EDUCATION Mechanical Engineering at the Politecnico di Milano, M.Sc. July 1998. Thesis: " Simulazione multibody mediante algebra dei quaternioni " (“Multibody simulation by means of quaternion algebra”). Italian State Certification for enabling the public profession in Engineering, 1999. ACADEMIC APPOINTMENTS Associate Professor, Università degli Studi di Parma, area ING-IND-13 09/A2 (Applied Mechanics), since 1/10/2014. Honorary Associate at the University of Wisconsin Madison, USA, since 2009. OTHER ACADEMIC TITLES AND ORGANIZING ACTIVITIES National Scientific Qualification ASN 2016, enabling to apply for a university Full Professor position. Deputy of the Università degli Studi di Parma at the ITS Maker Foundation, since 3/11/2016 Member of the Committee for the PhD in Industrial Engineering, at the Università degli Studi di Parma, since 2002. Member of the Committee for the assessment of PhD students in Industrial Engineering at the Università degli Studi di Parma, since 2013. Member of the Exams Committee and Opponent for the PhD theses at Technischen Universität Kaiserslautern, Germany (24/7/2015), Member of the Exams Committee and Opponent for the PhD theses at the Politecnico di Milano, Aerospace Engineering PhD (17/4/2009), Università degli Studi di Bergamo (14/4/2010, 26/4/2012), Università di Roma La Sapienza (9/11/2012), Politecnico di Milano, Mechanical Engineering (30/3/2015). -

BSM School Profile 2020
The British School of Milan www.britishschoolmilan.com Via Carlo Alberto Pisani Dossi, 16 - 20134 Milan, Italy Telephone: +39 02210941 CEEB Code: 748246 learning to excel since 1969 IB number: 3665 BSM SCHOOL PROFILE 720 3-18 40 100% Students Age Range IB Points Average A*- C IGCSE for Top 20 Students Grades The only school in Milan rated 100 43 “Excellent” by UK Government 50 107 Teachers Nationalities Inspectors Years of World-Class Co-curricular Education Activities Established 1969 Mission Independent, day school IB World School Our mission is to inspire learning within a caring, COBIS and HMC affiliated creative and international community, to pursue excellence, and to challenge students as they Not for Profit prepare for university and the world beyond. Rated One of the Top 10 Schools in Europe Graduating class of Average IB Diploma Score 100% 50 36 Points University Entry Assessment and Gr ading GCSE at age 16 • A* (highest) to G (lowest) • 9 (highest) to 1 (lowest) International Baccalaureate Diploma (IB) at age 18 • 6 subjects 7 (highest) - 1 (lowest) • Up to 3 Core points from Core (4,000-word Extended Essay + Theory of Knowledge) • Creativity, Activity, Service programme • Maximum 45 points The British School of Milan does not compute GPA or provide class ranking. Contact Information Primary Contact for University Enquiries Nicolas Amy Director of Sixth Form and University Counsellor [email protected] Chris Greenhalgh Principal and CEO [email protected] Julie Walker Head of Senior School [email protected] -

Heterogeneity of Colorectal Cancer Progression: Molecular Gas and Brakes
International Journal of Molecular Sciences Review Heterogeneity of Colorectal Cancer Progression: Molecular Gas and Brakes Federica Gaiani 1,2 , Federica Marchesi 3,4, Francesca Negri 5 , Luana Greco 6, Alberto Malesci 3,7, Gian Luigi de’Angelis 1,2 and Luigi Laghi 1,6,* 1 Department of Medicine and Surgery, University of Parma, 43126 Parma, Italy; [email protected] (F.G.); [email protected] (G.L.d.) 2 Gastroenterology and Endoscopy Unit, University-Hospital of Parma, via Gramsci 14, 43126 Parma, Italy 3 IRCCS Humanitas Research Hospital, via Manzoni 56, 20089 Rozzano, Italy; [email protected] (F.M.); [email protected] (A.M.) 4 Department of Medical Biotechnology and Translational Medicine, University of Milan, 20132 Milan, Italy 5 Medical Oncology Unit, University Hospital of Parma, 43126 Parma, Italy; [email protected] 6 Laboratory of Molecular Gastroenterology, IRCCS Humanitas Research Hospital, via Manzoni 56, 20089 Rozzano, Italy; [email protected] 7 Department of Biomedical Sciences, Humanitas University, Via Rita Levi Montalcini 4, 20072 Pieve Emanuele, Italy * Correspondence: [email protected] Abstract: The review begins with molecular genetics, which hit the field unveiling the involvement of oncogenes and tumor suppressor genes in the pathogenesis of colorectal cancer (CRC) and uncovering genetic predispositions. Then the notion of molecular phenotypes with different clinical behaviors was introduced and translated in the clinical arena, paving the way to next-generation sequencing that captured previously unrecognized heterogeneity. Among other molecular regulators of CRC progression, the extent of host immune response within the tumor micro-environment has a critical Citation: Gaiani, F.; Marchesi, F.; position. -

Third Mission at the Nursing Study Course University of Parma
Acta Biomed for Health Professions 2020; Vol. 91, S. 6: 125-127 DOI: 10.23750/abm.v91i6-S.10036 © Mattioli 1885 Focus on helthcare inter-professionals’ training Third mission at the Nursing Study course University of Parma. Intervention report Sandrino Luigi Marra1, Pasquale La Torre1, Michele Minari1, Giulia Pelosi1, Chiara Taffurelli1, Rita Romano1, Giuseppe Marletta1, Cristina Casubolo1, Margherita De Fanti2, Rachele La Sala1 1University Teaching Hospital, Parma, Italy; 2Indipendent researcher Abstract. Background: The term Third Mission refers to the activities with which universities interact directly with the communities and the territory of reference, combining the objectives of the third mission with the two traditional missions: teaching and research. These were the premises that guided the Nursing Studies Course of the University of Parma, with the goal of implementing a structured path of “Intercultural Nursing” on a demo-ethno-anthropological basis. Methods: The path taken was divided into several phases: arrangement of moments of interaction with students; teaching activity aimed at students based on the relationship between ethno-anthropological knowledge, aspects of migration medicine and social legislation; direct meetings with representatives of some communities present in the Parma area. Results: The interaction with students was achieved through meetings called “Cultural Coffee”. The first meetings, in the measure of 4-5, took place in the period October-December 2013, subsequently, the same number of meetings was repeated in the following years. In 2019, two important results were achieved: the creation of an ADE (teaching chosen by the student) dedicated to multiculturality. Another activity of the “Intercultural Nursing” course involve the students with some communities in the Parma area. -

Italian University Collections: Managing the Artistic Heritage of the University’S Ivory Tower
ENCATC JOURNAL OF CULTURAL MANAGEMENT & POLICY || Vol. 8, Issue 1, 2018 || ISSN 2224-2554 Italian university collections: managing the artistic heritage of the university’s ivory tower Isabella Mozzoni University of Parma, Italy [email protected] Simone Fanelli University of Parma, Italy [email protected] Chiara Carolina Donelli University of Parma, Italy [email protected] Submission date: 25.04.2018 • Acceptance date: 12.06.2018 • Publication date: 18.12.2018 ABSTRACT The management of university museums and collections has been an issue for decades as they have played a crucial role in supporting the three missions of the higher education system: research, teaching and making academia’s resources available Keywords: for public use. In this paper, we focus on the Italian case, where the enhancement, management and accessibility of university collections are all part of the evaluation Cultural system for universities. Our aim in this work is to propose a reconnaissance of university management art collections in Italy and investigate the three managerial challenges defined by the Council of Europe: accessibility, financial sustainability and communication of university University collections. The findings show that Italian universities hold an enormous cultural collection heritage, mainly undervalued, both in terms of number of artworks and in terms of the artworks’ economic value. In addition, Italian managerial approaches show significant University critical issues regarding the three managerial challenges. museum Artistic heritage ACKNOWLEDGEMENTS We would like to thank the attendees of the Sole 24 Ore Business School for their help in developing the questionnaire and collecting the data. Our sincere gratitude also goes to the museum services managers of the various universities involved in this study. -

For Peer Review Only
BMJ Open BMJ Open: first published as 10.1136/bmjopen-2015-009669 on 31 March 2016. Downloaded from Intravesical administration of combined hyaluronic acid (HA) and chondroitin sulphate (CS) for the treatment of female recurrent urinary tract infections: a European multicenter nested case-control study For peer review only Journal: BMJ Open Manuscript ID: bmjopen-2015-009669 Article Type: Research Date Submitted by the Author: 10-Aug-2015 Complete List of Authors: Ciani, Oriana; University of Exeter , University of Exeter Medical School; CeRGAS Bocconi, Arendsen, Erik; Diaconessenhuis, Dept. of Urology Romancik, Martin; St. Cyril and Method University Hospital, Dept. of Urology Lunik, Richard; Fakultná nemocnica s poliklinikou, Dept. of Urology Costantini, Elisabetta; University of Perugia, Dept. of Surgical and Biomedical Science Di Biase, Manuel; University of Perugia, Dept. of Surgical and Biomedical Science Morgia, Giuseppe; University of Catania, Dept. of Urology http://bmjopen.bmj.com/ Fragala', Eugenia; University of Catania, Dept. of Urology Tomaskin, Roman; Jessenius School of Medicine, Dept. of Urology Bernat, Marian; Fakultná nemocnica s poliklinikou, Dept. of Urology Guazzoni, Giorgio; Humanitas Clinical and Research Center, Dept. of Urology Tarricone, Rosanna; Bocconi University, Dept. of Policy Analysis and Public Management Lazzeri, Massimo; Humanitas Clinical and Research Center, Dept. of Urology on September 29, 2021 by guest. Protected copyright. <b>Primary Subject Urology Heading</b>: Secondary Subject Heading: Infectious diseases Urinary tract infections < UROLOGY, Antimicrobial Resistance, Hyaluronic Keywords: acid, chondroitin sulphate For peer review only - http://bmjopen.bmj.com/site/about/guidelines.xhtml Page 1 of 25 BMJ Open BMJ Open: first published as 10.1136/bmjopen-2015-009669 on 31 March 2016.