Evidence for a Protein-Protein Interaction Motif on an Acyl Carrier Protein Domain from a Modular Polyketide Synthase
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

In Vivo Evaluation of Ixabepilone (BMS247550), a Novel Epothilone B Derivative, Against Pediatric Cancer Models Jennifer K
Cancer Therapy: Preclinical In vivo Evaluation of Ixabepilone (BMS247550), A Novel Epothilone B Derivative, against Pediatric Cancer Models Jennifer K. Peterson,1Chandra Tucker,1Edward Favours,1PamelaJ. Cheshire,1Jeremy Creech,1 Catherine A. Billups,2 Richard Smykla,3 Francis Y.F. Lee,3 and Peter J. Houghton1 Abstract Purpose:Vinca alkaloids, agents that cause depolymerization of microtubules, are highly active in treatment of many pediatric cancers. In contrast, taxanes, agents that stabilize microtubules, are far less effective against the same cancer types.The purpose of the current study was to evaluate the antitumor activity of ixabepilone, an epothilone B derivative representing a new class of microtubule-stabilizing antimitotic agent in a wide variety of pediatric solid tumor models. Experimental Design: Ixabepilone was administered i.v. every 4 days for three doses to scid mice bearing s.c. human rhabdomyosarcoma (three lines), neuroblastoma (four),Wilms’ tumors (six), osteosarcoma (four), or brain tumors (seven).Tumor diameters were measured weekly, and tumor growth or regressions were determined. Pharmacokinetic studies were done following a single administration of drug at the maximum tolerated dose (MTD) level (10 mg/kg). Results: At the MTD (10 mg/kg), ixabepilone induced objective responses (all tumors in a group achieved z50% volume regression) in three of three rhabdomyosarcoma lines, three of five neuroblastomas, six of seven Wilms’ tumor models, two of six osteosarcoma, and four of eight brain tumor models. However, the dose-response curve was steep with only 2 of 19 tumors models regressing (z50%) at 4.4 mg/kg. In comparison, paclitaxel administered at the MTD on the same schedule failed to induce objective regressions of three tumor lines that were highly sensitive to treatment with ixabepilone. -

2701.Full-Text.Pdf
Cancer Therapy: Clinical The Effect of Ketoconazole on the Pharmacokinetics and Pharmacodynamics of Ixabepilone: A First in Class Epothilone B Analogue in Late-Phase Clinical Development Sanjay Goel,1Marvin Cohen,4 S. Nilgu« n C¸ o« mezoglu,4 Lionel Perrin,5 Franc¸ois Andre¤ ,5 DavidJayabalan, 1Lisa Iacono,4 Adriana Comprelli,4 Van T. Ly,4 Donglu Zhang,4 Carrie Xu,4 W. Griffith Humphreys,4 Hayley McDaid,1, 2 Gary Goldberg,1, 3 Susan B. Horwitz,1, 2 andSridhar Mani 1 Abstract Purpose:To determine if ixabepilone is a substrate for cytochrome P450 3A4 (CYP3A4) and if its metabolism by this cytochrome is clinically important, we did a clinical drug interaction study in humans using ketoconazole as an inhibitor of CYP3A4. Experimental Design: Human microsomes were usedto determine the cytochrome P450 enzyme(s) involvedin the metabolism of ixabepilone. Computational docking (CYP3A4) studies were done for epothilone B and ixabepilone. A follow-up clinical study was done in patients with cancer to determine if 400 mg/d ketoconazole (inhibitor of CYP3A4) altered the pharmacokinet- ics, drug-target interactions, and pharmacodynamics of ixabepilone. Results: Molecular modeling and human microsomal studies predicted ixabepilone to be a good substrate for CYP3A4. In patients, ketoconazole coadministration resulted in a maximum ixabepi- lone dose administration to 25 mg/m2 when comparedwith single-agent therapy of 40 mg/m 2. Coadministration of ketoconazole with ixabepilone resulted in a 79% increase in AUC0-1.The relationship of microtubule bundle formation in peripheral blood mononuclear cells to plasma ixabepilone concentration was well described by the Hill equation. Microtubule bundle formation in peripheral bloodmononuclear cells correlatedwith neutropenia. -

Contig Protein Description Symbol Anterior Posterior Ratio
Table S2. List of proteins detected in anterior and posterior intestine pooled samples. Data on protein expression are mean ± SEM of 4 pools fed the experimental diets. The number of the contig in the Sea Bream Database (http://nutrigroup-iats.org/seabreamdb) is indicated. Contig Protein Description Symbol Anterior Posterior Ratio Ant/Pos C2_6629 1,4-alpha-glucan-branching enzyme GBE1 0.88±0.1 0.91±0.03 0.98 C2_4764 116 kDa U5 small nuclear ribonucleoprotein component EFTUD2 0.74±0.09 0.71±0.05 1.03 C2_299 14-3-3 protein beta/alpha-1 YWHAB 1.45±0.23 2.18±0.09 0.67 C2_268 14-3-3 protein epsilon YWHAE 1.28±0.2 2.01±0.13 0.63 C2_2474 14-3-3 protein gamma-1 YWHAG 1.8±0.41 2.72±0.09 0.66 C2_1017 14-3-3 protein zeta YWHAZ 1.33±0.14 4.41±0.38 0.30 C2_34474 14-3-3-like protein 2 YWHAQ 1.3±0.11 1.85±0.13 0.70 C2_4902 17-beta-hydroxysteroid dehydrogenase 14 HSD17B14 0.93±0.05 2.33±0.09 0.40 C2_3100 1-acylglycerol-3-phosphate O-acyltransferase ABHD5 ABHD5 0.85±0.07 0.78±0.13 1.10 C2_15440 1-phosphatidylinositol phosphodiesterase PLCD1 0.65±0.12 0.4±0.06 1.65 C2_12986 1-phosphatidylinositol-4,5-bisphosphate phosphodiesterase delta-1 PLCD1 0.76±0.08 1.15±0.16 0.66 C2_4412 1-phosphatidylinositol-4,5-bisphosphate phosphodiesterase gamma-2 PLCG2 1.13±0.08 2.08±0.27 0.54 C2_3170 2,4-dienoyl-CoA reductase, mitochondrial DECR1 1.16±0.1 0.83±0.03 1.39 C2_1520 26S protease regulatory subunit 10B PSMC6 1.37±0.21 1.43±0.04 0.96 C2_4264 26S protease regulatory subunit 4 PSMC1 1.2±0.2 1.78±0.08 0.68 C2_1666 26S protease regulatory subunit 6A PSMC3 1.44±0.24 1.61±0.08 -
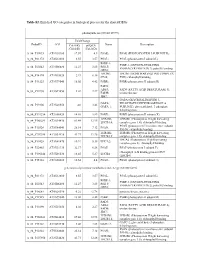
Table S2. Enriched GO Categories in Biological Process for the Shared Degs
Table S2. Enriched GO categories in biological process for the shared DEGs photosynthesis (GO ID:15979) Fold Change ProbeID AGI Col-0(R) pifQ(D) Name Description /Col-0(D) /Col-0(D) A_84_P19035 AT1G30380 17.07 4.9 PSAK; PSAK (PHOTOSYSTEM I SUBUNIT K) A_84_P21372 AT4G12800 8.55 3.57 PSAL; PSAL (photosystem I subunit L) PSBP-1; PSBP-1 (OXYGEN-EVOLVING A_84_P20343 AT1G06680 12.27 3.85 PSII-P; ENHANCER PROTEIN 2); poly(U) binding OEE2; LHCB6; LHCB6 (LIGHT HARVESTING COMPLEX A_84_P14174 AT1G15820 23.9 6.16 CP24; PSII); chlorophyll binding A_84_P11525 AT1G79040 16.02 4.42 PSBR; PSBR (photosystem II subunit R) FAD5; ADS3; FAD5 (FATTY ACID DESATURASE 5); A_84_P19290 AT3G15850 4.02 2.27 FADB; oxidoreductase JB67; GAPA (GLYCERALDEHYDE 3- GAPA; PHOSPHATE DEHYDROGENASE A A_84_P19306 AT3G26650 4.6 3.43 GAPA-1; SUBUNIT); glyceraldehyde-3-phosphate dehydrogenase A_84_P193234 AT2G06520 14.01 3.89 PSBX; PSBX (photosystem II subunit X) LHB1B1; LHB1B1 (Photosystem II light harvesting A_84_P160283 AT2G34430 89.44 32.95 LHCB1.4; complex gene 1.4); chlorophyll binding PSAN (photosystem I reaction center subunit A_84_P10324 AT5G64040 26.14 7.12 PSAN; PSI-N); calmodulin binding LHB1B2; LHB1B2 (Photosystem II light harvesting A_84_P207958 AT2G34420 41.71 12.26 LHCB1.5; complex gene 1.5); chlorophyll binding LHCA2 (Photosystem I light harvesting A_84_P19428 AT3G61470 10.91 5.36 LHCA2; complex gene 2); chlorophyll binding A_84_P22465 AT1G31330 32.37 6.58 PSAF; PSAF (photosystem I subunit F) chlorophyll A-B binding protein CP29 A_84_P190244 AT5G01530 16.45 5.27 LHCB4 -

Fatty Acid Biosynthesis
BI/CH 422/622 ANABOLISM OUTLINE: Photosynthesis Carbon Assimilation – Calvin Cycle Carbohydrate Biosynthesis in Animals Gluconeogenesis Glycogen Synthesis Pentose-Phosphate Pathway Regulation of Carbohydrate Metabolism Anaplerotic reactions Biosynthesis of Fatty Acids and Lipids Fatty Acids contrasts Diversification of fatty acids location & transport Eicosanoids Synthesis Prostaglandins and Thromboxane acetyl-CoA carboxylase Triacylglycerides fatty acid synthase ACP priming Membrane lipids 4 steps Glycerophospholipids Control of fatty acid metabolism Sphingolipids Isoprene lipids: Cholesterol ANABOLISM II: Biosynthesis of Fatty Acids & Lipids 1 ANABOLISM II: Biosynthesis of Fatty Acids & Lipids 1. Biosynthesis of fatty acids 2. Regulation of fatty acid degradation and synthesis 3. Assembly of fatty acids into triacylglycerol and phospholipids 4. Metabolism of isoprenes a. Ketone bodies and Isoprene biosynthesis b. Isoprene polymerization i. Cholesterol ii. Steroids & other molecules iii. Regulation iv. Role of cholesterol in human disease ANABOLISM II: Biosynthesis of Fatty Acids & Lipids Lipid Fat Biosynthesis Catabolism Fatty Acid Fatty Acid Degradation Synthesis Ketone body Isoprene Utilization Biosynthesis 2 Catabolism Fatty Acid Biosynthesis Anabolism • Contrast with Sugars – Lipids have have hydro-carbons not carbo-hydrates – more reduced=more energy – Long-term storage vs short-term storage – Lipids are essential for structure in ALL organisms: membrane phospholipids • Catabolism of fatty acids –produces acetyl-CoA –produces reducing -

Saturated Long-Chain Fatty Acid-Producing Bacteria Contribute
Zhao et al. Microbiome (2018) 6:107 https://doi.org/10.1186/s40168-018-0492-6 RESEARCH Open Access Saturated long-chain fatty acid-producing bacteria contribute to enhanced colonic motility in rats Ling Zhao1†, Yufen Huang2†, Lin Lu1†, Wei Yang1, Tao Huang1, Zesi Lin3, Chengyuan Lin1,4, Hiuyee Kwan1, Hoi Leong Xavier Wong1, Yang Chen5, Silong Sun2, Xuefeng Xie2, Xiaodong Fang2,5, Huanming Yang6, Jian Wang6, Lixin Zhu7* and Zhaoxiang Bian1* Abstract Background: The gut microbiota is closely associated with gastrointestinal (GI) motility disorder, but the mechanism(s) by which bacteria interact with and affect host GI motility remains unclear. In this study, through using metabolomic and metagenomic analyses, an animal model of neonatal maternal separation (NMS) characterized by accelerated colonic motility and gut dysbiosis was used to investigate the mechanism underlying microbiota-driven motility dysfunction. Results: An excess of intracolonic saturated long-chain fatty acids (SLCFAs) was associated with enhanced bowel motility in NMS rats. Heptadecanoic acid (C17:0) and stearic acid (C18:0), as the most abundant odd- and even- numbered carbon SLCFAs in the colon lumen, can promote rat colonic muscle contraction and increase stool frequency. Increase of SLCFAs was positively correlated with elevated abundances of Prevotella, Lactobacillus, and Alistipes. Functional annotation found that the level of bacterial LCFA biosynthesis was highly enriched in NMS group. Essential synthetic genes Fabs were largely identified from the genera Prevotella, Lactobacillus, and Alistipes. Pseudo germ-free (GF) rats receiving fecal microbiota from NMS donors exhibited increased defecation frequency and upregulated bacterial production of intracolonic SLCFAs. Modulation of gut dysbiosis by neomycin effectively attenuated GI motility and reduced bacterial SLCFA generation in the colon lumen of NMS rats. -

Crystal Structure of Fabz-ACP Complex Reveals a Dynamic Seesaw-Like Catalytic Mechanism of Dehydratase in Fatty Acid Biosynthesis
Cell Research (2016) 26:1330-1344. ORIGINAL ARTICLE www.nature.com/cr Crystal structure of FabZ-ACP complex reveals a dynamic seesaw-like catalytic mechanism of dehydratase in fatty acid biosynthesis Lin Zhang1, 2, Jianfeng Xiao3, Jianrong Xu1, Tianran Fu1, Zhiwei Cao1, Liang Zhu1, 2, Hong-Zhuan Chen1, 2, Xu Shen3, Hualiang Jiang3, Liang Zhang1, 2 1Department of Pharmacology and Chemical Biology, Shanghai Jiao Tong University School of Medicine, Shanghai, China; 2Shanghai Universities Collaborative Innovation Center for Translational Medicine, Shanghai, China; 3State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China Fatty acid biosynthesis (FAS) is a vital process in cells. Fatty acids are essential for cell assembly and cellular me- tabolism. Abnormal FAS directly correlates with cell growth delay and human diseases, such as metabolic syndromes and various cancers. The FAS system utilizes an acyl carrier protein (ACP) as a transporter to stabilize and shuttle the growing fatty acid chain throughout enzymatic modules for stepwise catalysis. Studying the interactions between enzymatic modules and ACP is, therefore, critical for understanding the biological function of the FAS system. How- ever, the information remains unclear due to the high flexibility of ACP and its weak interaction with enzymatic mod- ules. We present here a 2.55 Å crystal structure of type II FAS dehydratase FabZ in complex with holo-ACP, which exhibits a highly symmetrical FabZ hexamer-ACP3 stoichiometry with each ACP binding to a FabZ dimer subunit. Further structural analysis, together with biophysical and computational results, reveals a novel dynamic seesaw-like ACP binding and catalysis mechanism for the dehydratase module in the FAS system, which is regulated by a critical gatekeeper residue (Tyr100 in FabZ) that manipulates the movements of the β-sheet layer. -

Enabling Direct Compression Tablet Development Of
ENABLING DIRECT COMPRESSION TABLET DEVELOPMENT OF CELECOXIB THROUGH SOLID STATE ENGINEERING A DISSERTATION SUBMITTED TO THE FACULTY OF THE UNIVERSITY OF MINNESOTA BY Kunlin Wang IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY Changquan Calvin Sun, Advisor August 2020 © Kunlin Wang 2020 Acknowledgements Life as a Ph.D. student is a journey filled with gratitude. For my 5 years of Ph.D. study as well as 2 years of undergraduate research at the Sun Lab, the first person I would like to thank is my advisor, Prof. Changquan Calvin Sun. During my undergraduate research in my junior and senior years, he has been a good guide and scientist who triggered my interest in pharmaceutical materials science and lead my way to pursuing a Ph.D. degree in Pharmaceutics. During my graduate study, Dr. Sun has been a great mentor in science as well as a supporter who inspire me for attitude and solutions towards life’s problems. My appreciation also goes to my committee members, Dr. Raj Suryanarayanan, Dr. Timothy Wiedmann, and Dr. Andreas Stein for reviewing my dissertation, offering insightful comments and suggestions, and serving on my Ph. D. exam committees. I have learned the power of logical thinking and analysis from Dr. Sury; the significance of practical thinking from Dr. Wiedmann and the power of getting the big picture from Dr. Stein from a different perspective. I am extremely grateful not only for their time and patience, but for their trust in me as they offer me support in my development as a scientist. -

Medicinova Receives Notice of Allowance for Second Patent Covering MN-166 (Ibudilast) for the Treatment of Glioblastoma
MediciNova Receives Notice of Allowance for Second Patent Covering MN-166 (ibudilast) for the Treatment of Glioblastoma April 20, 2020 LA JOLLA, Calif., April 20, 2020 (GLOBE NEWSWIRE) -- MediciNova, Inc., a biopharmaceutical company traded on the NASDAQ Global Market (NASDAQ:MNOV) and the JASDAQ Market of the Tokyo Stock Exchange (Code Number: 4875), today announced that it has received a Notice of Allowance from the U.S. Patent and Trademark Office for a pending patent application which covers MN-166 (ibudilast) for the treatment of glioblastoma. This new patent has improved therapeutic claims compared to the first patent which covers MN-166 (ibudilast) for the treatment of glioblastoma, which was granted last year, and has a later expiration date than the first patent. Once issued, the patent maturing from this allowed patent application is expected to expire no earlier than February 2039. The allowed claims cover a method of treating a patient diagnosed with glioblastoma or recurrent glioblastoma, wherein the patient expresses methylated MGMT (O6-methylguanine-DNA methyltransferase), using MN-166 (ibudilast) in combination with one or more other therapeutic agents including temozolomide (TMZ), carmustine, bevacizumab, procarbazine, hydroxyurea, irinotecan, lomustine, nimotuzumab, sirolimus, mipsagargin, cabozantinib, onartuzumab, patupilone (epothilone B), and recombinant oncolytic poliovirus (PVS-RIPO). The allowed claims cover a wide range of doses of MN-166 (ibudilast) during an optionally repeating dosing cycle. The allowed claims also cover different types of glioblastoma including classical glioblastoma, proneural glioblastoma, mesenchymal glioblastoma, and neural glioblastoma. Yuichi Iwaki, MD, PhD, President and Chief Executive Officer of MediciNova, Inc., commented, "We are very pleased to receive notice that this new patent will be granted as it offers better coverage than our first patent covering glioblastoma. -

Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/Mtor Cascade Inhibitors: How Mutations Can Result in Therapy Resistance and How to Overcome Resistance
www.impactjournals.com/oncotarget/ Oncotarget, October, Vol.3, No 10 Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR Cascade Inhibitors: How Mutations Can Result in Therapy Resistance and How to Overcome Resistance James A. McCubrey1, Linda S. Steelman1, William H. Chappell1, Stephen L. Abrams1, Richard A. Franklin1, Giuseppe Montalto2, Melchiorre Cervello3, Massimo Libra4, Saverio Candido4, Grazia Malaponte4, Maria C. Mazzarino4, Paolo Fagone4, Ferdinando Nicoletti4, Jörg Bäsecke5, Sanja Mijatovic6, Danijela Maksimovic- Ivanic6, Michele Milella7, Agostino Tafuri8, Francesca Chiarini9, Camilla Evangelisti9, Lucio Cocco10, Alberto M. Martelli9,10 1 Department of Microbiology and Immunology, Brody School of Medicine at East Carolina University, Greenville, NC, USA 2 Department of Internal Medicine and Specialties, University of Palermo, Palermo, Italy 3 Consiglio Nazionale delle Ricerche, Istituto di Biomedicina e Immunologia Molecolare “Alberto Monroy”, Palermo, Italy 4 Department of Bio-Medical Sciences, University of Catania, Catania, Italy 5 Department of Medicine, University of Göttingen, Göttingen, Germany 6 Department of Immunology, Instititue for Biological Research “Sinisa Stankovic”, University of Belgrade, Belgrade, Serbia 7 Regina Elena National Cancer Institute, Rome, Italy 8 Sapienza, University of Rome, Department of Cellular Biotechnology and Hematology, Rome, Italy 9 Institute of Molecular Genetics, National Research Council-Rizzoli Orthopedic Institute, Bologna, Italy. 10 Department of Biomedical and Neuromotor Sciences, University of Bologna, Bologna, Italy Correspondence to: James A. McCubrey, email: [email protected] Keywords: Targeted Therapy, Therapy Resistance, Cancer Stem Cells, Raf, Akt, PI3K, mTOR Received: September 12, 2012, Accepted: October 18, 2012, Published: October 20, 2012 Copyright: © McCubrey et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. -

Interaction of Epothilone Analogs with the Paclitaxel Binding Site: Relationship Between Binding Affinity, Microtubule Stabilization, and Cytotoxicity
Chemistry & Biology, Vol. 11, 225–236, February, 2004, 2004 Elsevier Science Ltd. All rights reserved. DOI 10.1016/j.chembiol.2004.01.014 Interaction of Epothilone Analogs with the Paclitaxel Binding Site: Relationship between Binding Affinity, Microtubule Stabilization, and Cytotoxicity Rube´ n M. Buey,1 J. Fernando Dı´az,1,* sic microtubule-disrupting drugs (colchicine, vinblas- Jose´ M. Andreu,1 Aurora O’Brate,2 tine) block microtubule assembly by binding to the unas- Paraskevi Giannakakou,2 K.C. Nicolaou,3 sembled tubulin dimers and inactivating them, paclitaxel Pradip K. Sasmal,3 Andreas Ritze´ n,3 and docetaxel bind to microtubules, stabilizing them and Kenji Namoto3 and blocking their dynamics [2, 3]. Nevertheless, despite 1Centro de Investigaciones Biolo´ gicas their good activity against ovarian, metastatic breast, Consejo Superior de Investigaciones Cientı´ficas head and neck, and lung cancer [4], paclitaxel has two Ramiro de Maeztu 9 factors that hamper its applicability. First, its low aque- 28040 Madrid ous solubility, and second, the development of pleiotro- Spain pic drug resistance mediated both by the overexpres- 2 Winship Cancer Institute sion of the P-glycoprotein [5, 6] and the presence of Emory University School of Medicine mutations in -tubulin [7, 8]. Atlanta, Georgia 30322 The discovery in recent years of several natural sub- 3 Department of Chemistry and stances with a paclitaxel-like mechanism of action The Skaggs Institute for Chemical Biology (epothilone, discodermolide, laulimalide, eleutherobin, The Scripps Research Institute peloruside, dictyostatin-1, taccalonolide, and jatro- 10550 North Torrey Pines Road phane polyesters; [9–16]) opened new possibilities in La Jolla, California 92037 the field. -

Data-Driven Insights Into Ligands, Proteins, and Genetic Mutations
Data-Driven Insights into Ligands, Proteins, and Genetic Mutations by Jing Lu A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Bioinformatics) in the University of Michigan 2016 Doctoral Committee: Professor Heather A. Carlson, Chair Professor Charles L. Brooks III Assistant Professor Barry Grant Professor David S. Sept Professor Kerby A. Shedden © Jing Lu, 2016 Acknowledgements I would like to thank my advisor, Dr. Heather Carlson, for years of patient guidance, teaching, and support through the course of my PhD. I have learnt how to think critically and be rigorous in every step of research. I also want to express gratitude to my committee: Professor Charles L. Brooks III, Assistant Professor Barry Grant, Professor David S. Sept, Professor Kerby A. Shedden. Their advising is insightful and deepens my understanding of my research projects. I would like to thank Dr. Richard Smith for timely support for both my writing and research. For many Saturdays and Sundays, he promptly responds my requests for proofreading. Much of my work is built on his code in protein and ligand analysis. I would like to thank other members in Dr. Carlson’s lab for helping me with my work. Through the discussion with Dr. Jim Dunbar, I have learnt many critical ideas in Cheminformatics. Also, thank you to Sarah Graham and Jordan Clark for their tremendous friendship and willing to help with my writing. I would also thank previous members in Dr. Carlson’s lab. I would thank Dr. Phani Ghanakota for many late-night discussions and Dr.