Siponimod Enriches Regulatory T and B Lymphocytes in Secondary Progressive Multiple Sclerosis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Change of Fingolimod Patient Profiles Over Time
Journal of Personalized Medicine Article The Change of Fingolimod Patient Profiles over Time: A Descriptive Analysis of Two Non-Interventional Studies PANGAEA and PANGAEA 2.0 Tjalf Ziemssen 1,* and Ulf Schulze-Topphoff 2 1 Zentrum für Klinische Neurowissenschaften, Universitätsklinikum Carl Gustav Carus, D-01307 Dresden, Germany 2 Novartis Pharma GmbH, D-90429 Nuremberg, Germany; [email protected] * Correspondence: [email protected] Abstract: (1) Background: Fingolimod (Gilenya®) was the first oral treatment for patients with relapsing-remitting multiple sclerosis (RRMS). Since its approval, the treatment landscape has changed enormously. (2) Methods: Data of PANGAEA and PANGAEA 2.0, two German real-world studies, were descriptively analysed for possible evolution of patient profiles and treatment behavior. Both are prospective, multi-center, non-interventional, long-term studies on fingolimod use in RRMS in real life. Data of 4229 PANGAEA patients (recruited 2011–2013) and 2441 PANGAEA 2.0 patients (recruited 2015–2018) were available. Baseline data included demographics, RRMS characteristics and disease severity. (3) Results: The mean age of PANGAEA and PANGAEA 2.0 patients was similar (38.8 vs. 39.2 years). Patients in PANGAEA 2.0 had shorter disease duration (7.1 vs. 8.2 years) and fewer relapses in the year before baseline (1.2 vs. 1.6). Disease severity at baseline estimated by Citation: Ziemssen, T.; EDSS and SDMT was lower in PANGAEA 2.0 patients compared to PANGAEA (EDSS difference Schulze-Topphoff, U. The Change of 1.0 points; SDMT difference 3.3 points). (4) Conclusions: The results hint at an influence of changes in Fingolimod Patient Profiles over the treatment guidelines and the label on fingolimod patients profiles over time. -

Medical Review(S) Clinical Review
CENTER FOR DRUG EVALUATION AND RESEARCH APPLICATION NUMBER: 22-527 MEDICAL REVIEW(S) CLINICAL REVIEW Application Type NDA Application Number 22-527 Priority or Standard P Submit Date 12-21-2009 Received Date 12-21-2009 PDUFA Goal Date 9-21-2010 Division / Office FDA/ODE1 Reviewer Name Heather D. Fitter Review Completion Date August 26, 2010 Established Name Fingolimod (Proposed) Trade Name Gilenya Therapeutic Class S1P receptor modulator Applicant Novartis Formulation(s) Oral tablets Dosing Regimen 0.5 mg Indication(s) To reduce the frequency of relapses and delay the progression of disability in relapsing MS Intended Population(s) Relapsing Multiple Sclerosis Template Version: March 6, 2009 Clinical Review Heather D. Fitter, M.D. NDA 22-527 Fingolimod Table of Contents 1 RECOMMENDATIONS/RISK BENEFIT ASSESSMENT......................................... 7 1.1 Recommendation on Regulatory Action ............................................................. 7 1.2 Risk Benefit Assessment.................................................................................... 8 2 INTRODUCTION AND REGULATORY BACKGROUND ........................................ 8 2.1 Product Information ............................................................................................ 9 2.2 Table of Currently Available Treatments for the Proposed Indication................. 9 2.3 Availability of Proposed Active Ingredient in the United States ........................ 12 2.4 Important Safety Issues with Consideration to Related Drugs......................... -

Targeting Lysophosphatidic Acid in Cancer: the Issues in Moving from Bench to Bedside
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by IUPUIScholarWorks cancers Review Targeting Lysophosphatidic Acid in Cancer: The Issues in Moving from Bench to Bedside Yan Xu Department of Obstetrics and Gynecology, Indiana University School of Medicine, 950 W. Walnut Street R2-E380, Indianapolis, IN 46202, USA; [email protected]; Tel.: +1-317-274-3972 Received: 28 August 2019; Accepted: 8 October 2019; Published: 10 October 2019 Abstract: Since the clear demonstration of lysophosphatidic acid (LPA)’s pathological roles in cancer in the mid-1990s, more than 1000 papers relating LPA to various types of cancer were published. Through these studies, LPA was established as a target for cancer. Although LPA-related inhibitors entered clinical trials for fibrosis, the concept of targeting LPA is yet to be moved to clinical cancer treatment. The major challenges that we are facing in moving LPA application from bench to bedside include the intrinsic and complicated metabolic, functional, and signaling properties of LPA, as well as technical issues, which are discussed in this review. Potential strategies and perspectives to improve the translational progress are suggested. Despite these challenges, we are optimistic that LPA blockage, particularly in combination with other agents, is on the horizon to be incorporated into clinical applications. Keywords: Autotaxin (ATX); ovarian cancer (OC); cancer stem cell (CSC); electrospray ionization tandem mass spectrometry (ESI-MS/MS); G-protein coupled receptor (GPCR); lipid phosphate phosphatase enzymes (LPPs); lysophosphatidic acid (LPA); phospholipase A2 enzymes (PLA2s); nuclear receptor peroxisome proliferator-activated receptor (PPAR); sphingosine-1 phosphate (S1P) 1. -

The Role of Sirtuin 1 and Ceramide in T10c12 Conjugated Linoleic Acid
University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln Theses and Dissertations in Animal Science Animal Science Department Spring 4-2014 The Role of Sirtuin 1 and Ceramide in t10c12 Conjugated Linoleic Acid Induced Delipidation in 3T3-L1 Adipocytes Wei Wang University of Nebraska-Lincoln, [email protected] Follow this and additional works at: http://digitalcommons.unl.edu/animalscidiss Part of the Biochemical Phenomena, Metabolism, and Nutrition Commons, Biochemistry, Biophysics, and Structural Biology Commons, Biotechnology Commons, Comparative and Laboratory Animal Medicine Commons, and the Other Animal Sciences Commons Wang, Wei, "The Role of Sirtuin 1 and Ceramide in t10c12 Conjugated Linoleic Acid Induced Delipidation in 3T3-L1 Adipocytes" (2014). Theses and Dissertations in Animal Science. 81. http://digitalcommons.unl.edu/animalscidiss/81 This Article is brought to you for free and open access by the Animal Science Department at DigitalCommons@University of Nebraska - Lincoln. It has been accepted for inclusion in Theses and Dissertations in Animal Science by an authorized administrator of DigitalCommons@University of Nebraska - Lincoln. The Role of Sirtuin 1 and Ceramide in t10c12 Conjugated Linoleic Acid Induced Delipidation in 3T3-L1 Adipocytes By Wei Wang A DISSERTATION Presented to the Faculty of The Graduate College at the University of Nebraska In Partial Fulfillment of Requirements For the Degree of Doctor of Philosophy Major: Animal Science Under the Supervision of Professor Merlyn Nielsen Lincoln, Nebraska April, 2014 The Role of Sirtuin 1 and Ceramide in t10c12 Conjugated Linoleic Acid Induced Delipidation in 3T3-L1 Adipocytes Wei Wang, Ph.D. University of Nebraska, 2014 Advisers: Merlyn Nielsen and Michael Fromm Project 1: Trans-10, cis-12 conjugated linoleic acid (t10c12 CLA) reduces triglyceride (TG) levels in adipocytes through multiple pathways, with AMP-activated protein kinase (AMPK) generally facilitating, and peroxisome proliferator-activated receptor γ (PPARγ) generally opposing these reductions. -

Sphingolipids and Cell Signaling: Relationship Between Health and Disease in the Central Nervous System
Preprints (www.preprints.org) | NOT PEER-REVIEWED | Posted: 6 April 2021 doi:10.20944/preprints202104.0161.v1 Review Sphingolipids and cell signaling: Relationship between health and disease in the central nervous system Andrés Felipe Leal1, Diego A. Suarez1,2, Olga Yaneth Echeverri-Peña1, Sonia Luz Albarracín3, Carlos Javier Alméciga-Díaz1*, Angela Johana Espejo-Mojica1* 1 Institute for the Study of Inborn Errors of Metabolism, Faculty of Science, Pontificia Universidad Javeriana, Bogotá D.C., 110231, Colombia; [email protected] (A.F.L.), [email protected] (D.A.S.), [email protected] (O.Y.E.P.) 2 Faculty of Medicine, Universidad Nacional de Colombia, Bogotá D.C., Colombia; [email protected] (D.A.S.) 3 Nutrition and Biochemistry Department, Faculty of Science, Pontificia Universidad Javeriana, Bogotá D.C., Colombia; [email protected] (S.L.A.) * Correspondence: [email protected]; Tel.: +57-1-3208320 (Ext 4140) (C.J.A-D.). [email protected]; Tel.: +57-1-3208320 (Ext 4099) (A.J.E.M.) Abstract Sphingolipids are lipids derived from an 18-carbons unsaturated amino alcohol, the sphingosine. Ceramide, sphingomyelins, sphingosine-1-phosphates, gangliosides and globosides, are part of this group of lipids that participate in important cellular roles such as structural part of plasmatic and organelle membranes maintaining their function and integrity, cell signaling response, cell growth, cell cycle, cell death, inflammation, cell migration and differentiation, autophagy, angiogenesis, immune system. The metabolism of these lipids involves a broad and complex network of reactions that convert one lipid into others through different specialized enzymes. Impairment of sphingolipids metabolism has been associated with several disorders, from several lysosomal storage diseases, known as sphingolipidoses, to polygenic diseases such as diabetes and Parkinson and Alzheimer diseases. -

A Phase I Clinical Trial of Safingol in Combination with Cisplatin in Advanced Solid Tumors
Published OnlineFirst January 21, 2011; DOI: 10.1158/1078-0432.CCR-10-2323 Clinical Cancer Cancer Therapy: Clinical Research A Phase I Clinical Trial of Safingol in Combination with Cisplatin in Advanced Solid Tumors Mark A. Dickson1, Richard D. Carvajal1, Alfred H. Merrill, Jr.3, Mithat Gonen2, Lauren M. Cane1, and Gary K. Schwartz1 Abstract Purpose: Sphingosine 1-phosphate (S1P) is an important mediator of cancer cell growth and pro- liferation. Production of S1P is catalyzed by sphingosine kinase 1 (SphK). Safingol, (L-threo-dihydro- sphingosine) is a putative inhibitor of SphK. We conducted a phase I trial of safingol (S) alone and in combination with cisplatin (C). Experimental Design: A3þ 3 dose escalation was used. For safety, S was given alone 1 week before the combination. S þ C were then administered every 3 weeks. S was given over 60 to 120 minutes, depending on dose. Sixty minutes later, C was given over 60 minutes. The C dose of 75 mg/m2 was reduced in cohort 4 to 60 mg/m2 due to excessive fatigue. Results: Forty-three patients were treated, 41 were evaluable for toxicity, and 37 for response. The maximum tolerated dose (MTD) was S 840 mg/m2 over 120 minutes C 60 mg/m2, every 3 weeks. Dose- limiting toxicity (DLT) attributed to cisplatin included fatigue and hyponatremia. DLT from S was hepatic enzyme elevation. S pharmacokinetic parameters were linear throughout the dose range with no significant interaction with C. Patients treated at or near the MTD achieved S levels of more than 20 mmol/L and maintained levels greater than and equal to 5 mmol/L for 4 hours. -
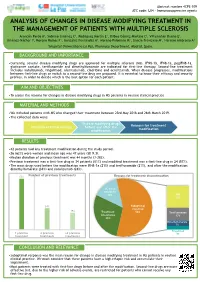
Analysis of Changes in Disease Modifying Treatment in The
Abstract number 4CPS-109 ATC code: L04 - Immunosuppressive agents ANALYSIS OF CHANGES IN DISEASE MODIFYING TREATMENT IN THE MANAGEMENT OF PATIENTS WITH MULTIPLE SCLEROSIS Arancón Pardo A1, Sobrino Jiménez C1, Rodríguez Martín E1, Bilbao Gómez-Martino C1, Villamañán Bueno E1, Jiménez-Nácher I1, Moreno Ramos F1, González Fernández A1, Moreno Palomino M1, García-Trevijano M1, Herrero Ambrosio A1 1Hospital Universitario La Paz, Pharmacy Department, Madrid, Spain. BACKGROUND AND IMPORTANCE •Currently, several disease modifying drugs are approved for multiple sclerosis (MS). IFNβ-1b, IFNβ-1a, pegIFNβ-1a, glatiramer acetate, teriflunomide and dimethylfumarate are indicated for first-line therapy. Second-line treatment includes natalizumab, fingolimod, alemtuzumab, cladribine and ocrelizumab. When disease progresses, modifications between first-line drugs or switch to a second-line drug are proposed. It is essential to know their efficacy and security profiles, in order to decide which is the best option for each patient. AIM AND OBJECTIVES •To assess the reasons for changes in disease modifying drugs in MS patients in routine clinical practice. MATERIAL AND METHODS •We included patients with MS who changed their treatment between 23rd May 2018 and 26th March 2019. •The collected data were: Disease modifying drugs Reasons for treatment Duration of initial therapy before and after the modification modification RESULTS •42 patients had any treatment modification during the study period. •26 (62%) were women and mean age was 47 years (SD 9.3). •Median duration of previous treatment was 44 months (3-282). •Previous treatment was a first-line drug in 34 patients (81%) and modified treatment was a first-line drug in 24 (57%). -

Patient Focused Disease State and Assistance Programs
Patient Focused Disease State and Assistance Programs Medication Medication Toll-free Brand (Generic) Website number Additional Resources Allergy/Asthma Xolair (omalizumab) xolair.com 1-866-4-XOLAIR lung.org Cardiovascular Pradaxa (dabigatran) pradaxa.com 877-481-5332 heart.org Praluent (alirocumab) praluent.com 844-PRALUENT thefhfoundation.org Repatha (evolocumab) repatha.com 844-REPATHA Tikosyn (dofetilide) tikosyn.com 800-879-3477 Crohn’s Disease Cimzia (certolizumab pegol) cimzia.com 866-4-CIMZIA crohnsandcolitis.com Humira (adalimumab) humira.com 800-4-HUMIRA crohnsforum.com Stelara (ustekinumab) stelarainfo.com 877-STELARA Dermatology Cosentyx (secukinumab) cosentyx.com 844-COSENTYX psoriasis.org Dupixent (dupilumab) dupixent.com 844-DUPIXENT nationaleczema.org Enbrel (etanercept) enbrel.com 888-4-ENBREL Humira (adalimumab) humira.com 800-4-HUMIRA Otezla (apremilast) otezla.com 844-4-OTEZLA Stelara (ustekinumab) stelarainfo.com 877-STELARA Taltz (ixekizumab) taltz.com 800-545-5979 Hematology Aranesp (darbepoetin alfa) aranesp.com 805-447-1000 chemocare.com Granix (filgrastim) granixrx.com 888-4-TEVARX hematology.org Jadenu (deferasirox) jadenu.com 888-282-7630 Neulasta (pegfilgrastim) neulasta.com 800-77-AMGEN Neupogen (filgrastim) neupogen.com 800-77-AMGEN Nivestym (filgrastim) nivestym.com 800-879-3477 Zarxio (filgrastim) zarxio.com 800-525-8747 Zytiga (abiraterone) zytiga.com 800-JANSSEN Hepatitis B Baraclude (entecavir) baraclude.com 800-321-1335 cdc.gov Viread (tenofovir disoproxil viread.com 800-GILEAD-5 hepb.org fumarate) -

COMPARISON of the WHO ATC CLASSIFICATION & Ephmra/Intellus Worldwide ANATOMICAL CLASSIFICATION
COMPARISON OF THE WHO ATC CLASSIFICATION & EphMRA/Intellus Worldwide ANATOMICAL CLASSIFICATION: VERSION June 2019 2 Comparison of the WHO ATC Classification and EphMRA / Intellus Worldwide Anatomical Classification The following booklet is designed to improve the understanding of the two classification systems. The development of the two systems had previously taken place separately. EphMRA and WHO are now working together to ensure that there is a convergence of the 2 systems rather than a divergence. In order to better understand the two classification systems, we should pay attention to the way in which substances/products are classified. WHO mainly classifies substances according to the therapeutic or pharmaceutical aspects and in one class only (particular formulations or strengths can be given separate codes, e.g. clonidine in C02A as antihypertensive agent, N02C as anti-migraine product and S01E as ophthalmic product). EphMRA classifies products, mainly according to their indications and use. Therefore, it is possible to find the same compound in several classes, depending on the product, e.g., NAPROXEN tablets can be classified in M1A (antirheumatic), N2B (analgesic) and G2C if indicated for gynaecological conditions only. The purposes of classification are also different: The main purpose of the WHO classification is for international drug utilisation research and for adverse drug reaction monitoring. This classification is recommended by the WHO for use in international drug utilisation research. The EphMRA/Intellus Worldwide classification has a primary objective to satisfy the marketing needs of the pharmaceutical companies. Therefore, a direct comparison is sometimes difficult due to the different nature and purpose of the two systems. -

The Real-World Patient Experience of Fingolimod and Dimethyl Fumarate
Wicks et al. BMC Res Notes (2016) 9:434 DOI 10.1186/s13104-016-2243-8 BMC Research Notes RESEARCH ARTICLE Open Access The real‑world patient experience of fingolimod and dimethyl fumarate for multiple sclerosis Paul Wicks1* , Lawrence Rasouliyan2, Bo Katic1, Beenish Nafees3, Emuella Flood3 and Rahul Sasané4 Abstract Background: Oral disease-modifying therapies offer equivalent or superior efficacy and greater convenience versus injectable options. Objectives: To compare patient-reported experiences of fingolimod and dimethyl fumarate. Methods: Adult relapsing-remitting multiple sclerosis patients treated with fingolimod or dimethyl fumarate were recruited from an online patient community and completed an online survey about treatment side effects, discon- tinuation, and satisfaction. Results: 281 patients in four groups completed the survey: currently receiving fingolimod (CF, N 61), currently receiving dimethyl fumarate (CDMF, N 129), discontinued fingolimod (DF, N 32) and discontinued= dimethyl fuma- rate (DDMF, N 59). Reasons for treatment= switch were to take oral treatment =(CF: 63.3 %, CDMF: 61.8 %), side effects of prior medication= (CF: 67.3 %, CDMF: 44.1 %) and lack of effectiveness of prior medication (CF: 38.8 %, CDMF: 31.4 %). Main reasons for discontinuation were side effects (DF: 46.9 %, DDMF: 67.8 %) and lack of effectiveness (DF: 25.0 %, DDMF: 15.3 %). CDMF patients had an increased risk of abdominal pain, flushing, diarrhea, and nausea. Treatment satisfaction was highest among CF patients followed by CDMF, DF, and then DDMF patients. Conclusions: Discontinuation was driven by experience of side effects. Patients currently taking dimethyl fumarate were more likely to experience a side effect versus patients currently taking fingolimod. -

Fingolimod (Gilenya)
Fingolimod (Gilenya) This factsheet is about fingolimod, a Whether you’ll be offered this or any other DMT disease modifying therapy (DMT) for depends on whether you qualify for it based relapsing multiple sclerosis (MS). on guidelines used by your neurologist. These come from NICE and the Association of British At the end of this factsheet you’ll find out where Neurologists and are based on a drug’s Europe- you can get more information on this drug, other wide licence. drugs for MS and the benefits of early treatment. In England there are also rules from NHS England about who can have the different DMTs and when. This factsheet doesn’t cover everything about Scotland, Wales and Northern Ireland also have this drug and shouldn’t be used in place of their own guidelines for many DMTs. advice from your MS specialist team. For more information speak to them and read the online information from the drug’s makers (see the Whether you can have a drug also depends on section More information and support). if the NHS where you live will pay for it. NHS England guidelines on this tend to follow what What is fingolimod? NICE says. Fingolimod is a drug that was first given a licence These people can have fingolimod: to be used against relapsing MS in the UK in 2011. In England and Northern Ireland: In 2012 the National Institute for Health and Care Excellence (NICE) gave the go ahead for it to be • People with ‘highly active relapsing remitting used on the NHS. -

Targeting the Sphingosine Kinase/Sphingosine-1-Phosphate Signaling Axis in Drug Discovery for Cancer Therapy
cancers Review Targeting the Sphingosine Kinase/Sphingosine-1-Phosphate Signaling Axis in Drug Discovery for Cancer Therapy Preeti Gupta 1, Aaliya Taiyab 1 , Afzal Hussain 2, Mohamed F. Alajmi 2, Asimul Islam 1 and Md. Imtaiyaz Hassan 1,* 1 Centre for Interdisciplinary Research in Basic Sciences, Jamia Millia Islamia, Jamia Nagar, New Delhi 110025, India; [email protected] (P.G.); [email protected] (A.T.); [email protected] (A.I.) 2 Department of Pharmacognosy, College of Pharmacy, King Saud University, Riyadh 11451, Saudi Arabia; afi[email protected] (A.H.); [email protected] (M.F.A.) * Correspondence: [email protected] Simple Summary: Cancer is the prime cause of death globally. The altered stimulation of signaling pathways controlled by human kinases has often been observed in various human malignancies. The over-expression of SphK1 (a lipid kinase) and its metabolite S1P have been observed in various types of cancer and metabolic disorders, making it a potential therapeutic target. Here, we discuss the sphingolipid metabolism along with the critical enzymes involved in the pathway. The review provides comprehensive details of SphK isoforms, including their functional role, activation, and involvement in various human malignancies. An overview of different SphK inhibitors at different phases of clinical trials and can potentially be utilized as cancer therapeutics has also been reviewed. Citation: Gupta, P.; Taiyab, A.; Hussain, A.; Alajmi, M.F.; Islam, A.; Abstract: Sphingolipid metabolites have emerged as critical players in the regulation of various Hassan, M..I. Targeting the Sphingosine Kinase/Sphingosine- physiological processes. Ceramide and sphingosine induce cell growth arrest and apoptosis, whereas 1-Phosphate Signaling Axis in Drug sphingosine-1-phosphate (S1P) promotes cell proliferation and survival.