Chemical Strategies for Investigation of Deubiquitinases
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Supporting Information
Supporting Information Figure S1. The functionality of the tagged Arp6 and Swr1 was confirmed by monitoring cell growth and sensitivity to hydeoxyurea (HU). Five-fold serial dilutions of each strain were plated on YPD with or without 50 mM HU and incubated at 30°C or 37°C for 3 days. Figure S2. Localization of Arp6 and Swr1 on chromosome 3. The binding of Arp6-FLAG (top), Swr1-FLAG (middle), and Arp6-FLAG in swr1 cells (bottom) are compared. The position of Tel 3L, Tel 3R, CEN3, and the RP gene are shown under the panels. Figure S3. Localization of Arp6 and Swr1 on chromosome 4. The binding of Arp6-FLAG (top), Swr1-FLAG (middle), and Arp6-FLAG in swr1 cells (bottom) in the whole chromosome region are compared. The position of Tel 4L, Tel 4R, CEN4, SWR1, and RP genes are shown under the panels. Figure S4. Localization of Arp6 and Swr1 on the region including the SWR1 gene of chromosome 4. The binding of Arp6- FLAG (top), Swr1-FLAG (middle), and Arp6-FLAG in swr1 cells (bottom) are compared. The position and orientation of the SWR1 gene is shown. Figure S5. Localization of Arp6 and Swr1 on chromosome 5. The binding of Arp6-FLAG (top), Swr1-FLAG (middle), and Arp6-FLAG in swr1 cells (bottom) are compared. The position of Tel 5L, Tel 5R, CEN5, and the RP genes are shown under the panels. Figure S6. Preferential localization of Arp6 and Swr1 in the 5′ end of genes. Vertical bars represent the binding ratio of proteins in each locus. -

Supplementary Tables
Supplementary Tables Supplementary Table S1: Preselected miRNAs used in feature selection Univariate Cox proportional hazards regression analysis of the endpoint freedom from recurrence in the training set (DKTK-ROG sample) allowed the pre-selection of 524 miRNAs (P< 0.5), which were used in the feature selection. P-value was derived from log-rank test. miRNA p-value miRNA p-value miRNA p-value miRNA p-value hsa-let-7g-3p 0.0001520 hsa-miR-1304-3p 0.0490161 hsa-miR-7108-5p 0.1263245 hsa-miR-6865-5p 0.2073121 hsa-miR-6825-3p 0.0004257 hsa-miR-4298 0.0506194 hsa-miR-4453 0.1270967 hsa-miR-6893-5p 0.2120664 hsa-miR-668-3p 0.0005188 hsa-miR-484 0.0518625 hsa-miR-200a-5p 0.1276345 hsa-miR-25-3p 0.2123829 hsa-miR-3622b-3p 0.0005885 hsa-miR-6851-3p 0.0531446 hsa-miR-6090 0.1278692 hsa-miR-3189-5p 0.2136060 hsa-miR-6885-3p 0.0006452 hsa-miR-1276 0.0557418 hsa-miR-148b-3p 0.1279811 hsa-miR-6073 0.2139702 hsa-miR-6875-3p 0.0008188 hsa-miR-3173-3p 0.0559962 hsa-miR-4425 0.1288330 hsa-miR-765 0.2141536 hsa-miR-487b-5p 0.0011381 hsa-miR-650 0.0564616 hsa-miR-6798-3p 0.1293342 hsa-miR-338-5p 0.2153079 hsa-miR-210-5p 0.0012316 hsa-miR-6133 0.0571407 hsa-miR-4472 0.1300006 hsa-miR-6806-5p 0.2173515 hsa-miR-1470 0.0012822 hsa-miR-4701-5p 0.0571720 hsa-miR-4465 0.1304841 hsa-miR-98-5p 0.2184947 hsa-miR-6890-3p 0.0016539 hsa-miR-202-3p 0.0575741 hsa-miR-514b-5p 0.1308790 hsa-miR-500a-3p 0.2185577 hsa-miR-6511b-3p 0.0017165 hsa-miR-4733-5p 0.0616138 hsa-miR-378c 0.1317442 hsa-miR-4515 0.2187539 hsa-miR-7109-3p 0.0021381 hsa-miR-595 0.0629350 hsa-miR-3121-3p -

(Ubl-Ptms): Small Peptides with Huge Impact in Liver Fibrosis
cells Review Ubiquitin-Like Post-Translational Modifications (Ubl-PTMs): Small Peptides with Huge Impact in Liver Fibrosis 1 1, 1, Sofia Lachiondo-Ortega , Maria Mercado-Gómez y, Marina Serrano-Maciá y , Fernando Lopitz-Otsoa 2, Tanya B Salas-Villalobos 3, Marta Varela-Rey 1, Teresa C. Delgado 1,* and María Luz Martínez-Chantar 1 1 Liver Disease Lab, CIC bioGUNE, Centro de Investigación Biomédica en Red de Enfermedades Hepáticas y Digestivas (CIBERehd), 48160 Derio, Spain; [email protected] (S.L.-O.); [email protected] (M.M.-G.); [email protected] (M.S.-M.); [email protected] (M.V.-R.); [email protected] (M.L.M.-C.) 2 Liver Metabolism Lab, CIC bioGUNE, 48160 Derio, Spain; fl[email protected] 3 Department of Biochemistry and Molecular Medicine, School of Medicine, Autonomous University of Nuevo León, Monterrey, Nuevo León 66450, Mexico; [email protected] * Correspondence: [email protected]; Tel.: +34-944-061318; Fax: +34-944-061301 These authors contributed equally to this work. y Received: 6 November 2019; Accepted: 1 December 2019; Published: 4 December 2019 Abstract: Liver fibrosis is characterized by the excessive deposition of extracellular matrix proteins including collagen that occurs in most types of chronic liver disease. Even though our knowledge of the cellular and molecular mechanisms of liver fibrosis has deeply improved in the last years, therapeutic approaches for liver fibrosis remain limited. Profiling and characterization of the post-translational modifications (PTMs) of proteins, and more specifically NEDDylation and SUMOylation ubiquitin-like (Ubls) modifications, can provide a better understanding of the liver fibrosis pathology as well as novel and more effective therapeutic approaches. -

The Effect of the URM1 Pathway on Translation by Thiolation of Specific Trnas
Research Collection Doctoral Thesis The effect of the URM1 pathway on translation by thiolation of specific tRNAs Author(s): Rezgui, Vanessa Anissa Nathalie Publication Date: 2012 Permanent Link: https://doi.org/10.3929/ethz-a-007624233 Rights / License: In Copyright - Non-Commercial Use Permitted This page was generated automatically upon download from the ETH Zurich Research Collection. For more information please consult the Terms of use. ETH Library DISS. ETH NO. 20870 The effect of the URM1 pathway on translation by thiolation of specific tRNAs A dissertation submitted to ETH ZURICH for the degree of Doctor of Sciences presented by Vanessa Anissa Nathalie Rezgui M.Sc. University of Geneva born on April, 28th 1981 citizen of Switzerland (Zurich) accepted on the recommendation of Prof. Matthias Peter – examiner Prof. André Gerber – co-examiner Prof. Paola Picotti – co-examiner 2012 TABLE OF CONTENTS SUMMARY ........................................................................................................ 4 RESUMÉ ............................................................................................................ 5 1 Introduction. ............................................................................................... 6 1.1 Translation............................................................................................................................................ 6 1.2 Translation regulation...................................................................................................................... -

Urm1 in Trna Thiolation and Protein Modification
Urm1 in tRNA thiolation and protein modification Annemarthe G. van der Veen Urm1 in tRNA thiolation and protein modification A.G. van der Veen Thesis, Utrecht University, The Netherlands A.G. van der Veen 2011 ISBN: 9789461081728 Cover image: Stata Center, MIT campus. Photo courtesy of W. van der Veen Invitation: Image courtesy of Eugene Galleries, Boston Printed by: Gildeprint Drukkerijen, Enschede Urm1 in tRNA thiolation and protein modification Urm1 in tRNA thiolatie en eiwit modificatie (met een samenvatting in het Nederlands) Proefschrift ter verkrijging van de graad van doctor aan de Universiteit Utrecht op gezag van de rector magnificus, prof.dr. G.J. van der Zwaan, ingevolge het besluit van het college voor promoties in het openbaar te verdedigen op maandag 30 mei 2011 des middags te 12.45 uur door Annemarthe Godelieve van der Veen geboren op 13 september 1982 te Hardenberg Promotoren: Prof.dr. H.L. Ploegh Prof.dr. E.J.H.J. Wiertz The research described in this thesis was supported by a pre-doctoral grant from the Boehringer Ingelheim Fonds. Voor papa en mama Contents Chapter 1 General introduction 8 Chapter 2 A functional proteomics approach links the Ubiquitin- 28 related modifier Urm1 to a tRNA modification pathway Chapter 3 Role of the Ubiquitin-like molecule Urm1 as a 46 noncanonical lysine-directed protein modifier Chapter 4 Transgenic mice expressing a dominant negative Urm1 74 mutant fail to generate offspring Chapter 5 Urm1B, an alternate isoform of Urm1, is stabilized and 90 redistributed by its interaction with ATPBD3 Chapter 6 General discussion 102 English summary 114 Nederlandse samenvatting 116 Acknowledgements 118 Curriculum vitae 121 List of publications 122 Chapter 1 General introduction Post-translational modifications Cellular homeostasis requires tight control of protein activity, function, stability, and localization. -

Urm1 Couples Sulfur Transfer to Ubiquitin-Like Protein Function in Oxidative Stress
COMMENTARY Urm1 couples sulfur transfer to ubiquitin-like protein function in oxidative stress Matthew D. Petroskia, Guy S. Salvesenb, and Dieter A. Wolfa,1 aSignal Transduction Program and bProgram in Apoptosis and Cell Death Research, Sanford-Burnham Medical Research Institute, La Jolla, CA 92037 he posttranslational modification of proteins with ubiquitin and T ubiquitin-like proteins (collec- tively referred to as UBLs) has emerged as a major regulatory mechanism in eukaryotes. UBLs are characterized by a core β-grasp fold and an essential car- boxy terminal glycine within a di-glycine motif (1). These features are also found in several prokaryotic sulfur carriers, sug- gestive of an evolutionary relationship to UBLs (2–5). A case in point is the UBL Urm1 (ubiquitin-related modifier 1). Urm1 is known to be conjugated to the peroxiredoxin Ahp1 (6), but its sequence and structure more closely resembles bacterial sulfur carrier proteins (7). In PNAS, work by Van der Veen et al. pro- vides important insight into the mecha- nism of Urm1 conjugation that highlights its position as an ancient UBL in eukar- yotes and solidifies its roles as a post- translational protein modifier involved in oxidative stress response mechanisms (8). All UBLs require ATP-dependent acti- Fig. 1. Juxtaposition of Urm1 as a prokaryotic sulfur carrier and a eukaryotic UBL. The shaded areas vation through their cognate E1 enzyme indicate overlapping aspects of the pathways, illustrating that Urm1 seems to use features common to both ubiquitin and MoaD for its activation and conjugation. The Urm1–E2 intermediate indicated in before conjugation onto target proteins brackets is still hypothetical but may account for the hydroxylamine sensitivity of Urm1–protein con- (Fig. -

The E3 Ligase TRIM32 Is an Effector of the RAS Family Gtpase RAP2
The E3 Ligase TRIM32 is an effector of the RAS family GTPase RAP2 Berna Demiray A thesis submitted towards the degree of Doctor of Philosophy Cancer Institute University College London 2014 Declaration I, Berna Demiray, confirm that the work presented in this thesis is my own. Where information has been derived from other sources, I confirm that this has been indicated. London, 2014 The E3 Ligase TRIM32 is an Effector of the RAS family GTPase RAP2 Classical RAS oncogenes are mutated in approximately 30% of human tumours and RAP proteins are closely related to classical RAS proteins. RAP1 has an identical effector domain to RAS whereas RAP2 differs by one amino acid. RAP2 not only shares effectors with other classical RAS family members, but it also has its own specific effectors that do not bind to RAP1 or classical RAS family proteins. Thus, although closely related, RAP2 performs distinct functions, although these have been poorly characterised. Using RAP2 as bait in Tandem Affinity Purifications, we have identified several RAP2 interacting proteins including TRIM32; a protein implicated in diverse pathological processes such as Limb-Girdle Muscular Dystrophy (LGMD2H), and Bardet-Biedl syndrome (BBS). TRIM32 was shown to interact specifically with RAP2 in an activation- and effector domain-dependent manner; demonstrating stronger interaction with the RAP2 V12 mutant than the wild-type RAP2 and defective binding to the effector mutant RAP2 V12A38. The interaction was mapped to the C-terminus of TRIM32 (containing the NHL domains) while mutations found in LGMD2H (R394H, D487N, ∆588) were found to disrupt binding to RAP2. The TRIM32 P130S mutant linked to BBS did not affect binding to RAP2, suggesting that the RAP2-TRIM32 interaction may be functionally involved in LGMD2H. -

Urmylation and Trna Thiolation Functions of Ubiquitinlike Uba4urm1
FEBS Letters 589 (2015) 904–909 journal homepage: www.FEBSLetters.org Urmylation and tRNA thiolation functions of ubiquitin-like Uba4ÁUrm1 systems are conserved from yeast to manq André Jüdes a, Folke Ebert a, Christian Bär a,b, Kathrin L. Thüring c, Aileen Harrer a, Roland Klassen a, ⇑ Mark Helm c, Michael J.R. Stark d, Raffael Schaffrath a, a Universität Kassel, Institut für Biologie, FG Mikrobiologie, Heinrich-Plett-Str. 40, 34132 Kassel, Germany b Spanish National Cancer Centre, Melchor Fernandez Almagro 3, Madrid, Spain c Johannes Gutenberg Universität Mainz, Institut für Pharmazie und Biochemie, Staudinger Weg 5, 55128 Mainz, Germany d Centre for Gene Regulation & Expression, College of Life Sciences, MSI/WTB Complex, University of Dundee, Dundee DD1 5EH, Scotland, UK article info abstract Article history: The ubiquitin-like protein Urm1 from budding yeast and its E1-like activator Uba4 have dual roles in Received 12 January 2015 protein urmylation and tRNA thiolation pathways. To study whether these are conserved among Revised 11 February 2015 eukaryotes, we used gene shuffles to replace the yeast proteins by their human counterparts, Accepted 24 February 2015 hURM1 and hUBA4/MOCS3. As judged from biochemical and genetical assays, hURM1 and hUBA4 Available online 3 March 2015 are functional in yeast, albeit at reduced efficiencies. They mediate urmylation of the peroxiredoxin Edited by Michael Ibba Ahp1, a known urmylation target in yeast, and support tRNA thiolation. Similar to hUBA4, yeast Uba4 itself is modified by Urm1 and hURM1 suggesting target overlap between eukaryal urmylation pathways. In sum, our study shows that dual-function ubiquitin-like Urm1 Uba4 systems are con- Keywords: Á Urm1 (hURM1) served and exchangeable between human and yeast cells. -

Architecture of the Yeast Elongator Complex M Dauden, J Kosinski, Olga Kolaj-Robin, a Desfosses, a Ori, C Faux, N Hoffmann, O Onuma, K Breunig, M Beck, Etal
Architecture of the yeast elongator complex M Dauden, J Kosinski, Olga Kolaj-Robin, A Desfosses, A Ori, C Faux, N Hoffmann, O Onuma, K Breunig, M Beck, etal. To cite this version: M Dauden, J Kosinski, Olga Kolaj-Robin, A Desfosses, A Ori, et al.. Architecture of the yeast elon- gator complex. EMBO Reports, EMBO Press, 2017, 18 (2), pp.264-279. 10.15252/embr.201643353. hal-02289580 HAL Id: hal-02289580 https://hal.archives-ouvertes.fr/hal-02289580 Submitted on 16 Sep 2019 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Article Architecture of the yeast Elongator complex Maria I Dauden1, Jan Kosinski1, Olga Kolaj-Robin2,3,4, Ambroise Desfosses1,†, Alessandro Ori1,‡, Celine Faux2,3,4,§, Niklas A Hoffmann1, Osita F Onuma5, Karin D Breunig5 , Martin Beck1 , Carsten Sachse1 , Bertrand Séraphin2,3,4, Sebastian Glatt6,* & Christoph W Müller1,** Abstract chaperones [1–3]. Previous studies indicated that specific base modifications in the wobble base position of tRNAs are crucial to The highly conserved eukaryotic Elongator complex performs maintain these highly dynamic and complex mechanisms. Mainly specific chemical modifications on wobble base uridines of tRNAs, because they influence the recognition rate and affinity between which are essential for proteome stability and homeostasis. -

Title: a Yeast Phenomic Model for the Influence of Warburg Metabolism on Genetic
bioRxiv preprint doi: https://doi.org/10.1101/517490; this version posted January 15, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. 1 Title Page: 2 3 Title: A yeast phenomic model for the influence of Warburg metabolism on genetic 4 buffering of doxorubicin 5 6 Authors: Sean M. Santos1 and John L. Hartman IV1 7 1. University of Alabama at Birmingham, Department of Genetics, Birmingham, AL 8 Email: [email protected], [email protected] 9 Corresponding author: [email protected] 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 1 bioRxiv preprint doi: https://doi.org/10.1101/517490; this version posted January 15, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. 26 Abstract: 27 Background: 28 Saccharomyces cerevisiae represses respiration in the presence of adequate glucose, 29 mimicking the Warburg effect, termed aerobic glycolysis. We conducted yeast phenomic 30 experiments to characterize differential doxorubicin-gene interaction, in the context of 31 respiration vs. glycolysis. The resulting systems level biology about doxorubicin 32 cytotoxicity, including the influence of the Warburg effect, was integrated with cancer 33 pharmacogenomics data to identify potentially causal correlations between differential 34 gene expression and anti-cancer efficacy. -
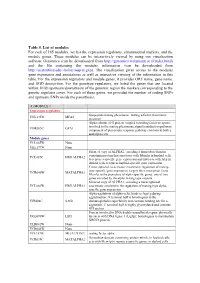
Table 5. List of Modules for Each of 165 Modules, We List the Expression Regulators, Chromosomal Markers, and the Module Genes
Table 5. List of modules For each of 165 modules, we list the expression regulators, chromosomal markers, and the module genes. These modules can be interactively viewed by using our visualization software Genomica (can be downloaded from http://genomica.weizmann.ac.il/index.html) and the file containing the modules information (can be downloaded from http://ai.stanford.edu/~koller/seqvar.gxp). The visualization gives access to the modules gene expression and annotations as well as interactive viewing of the information in this table. For the expression regulators and module genes, it provides ORF name, gene name, and SGD desciprtion. For the genotype regulators, we listed the genes that are located within 10 kb upstream/downstream of the genomic region the markers corresponding to the genetic regulator cover. For each of these genes, we provided the number of coding SNPs and upstream SNPs inside the parenthesis. (1) MODULE 1 Expression regulators lipopeptide mating pheromone; mating a-factor pheromone YNL145W MFA2 precursor Alpha subunit of G protein coupled to mating factor receptors, involved in the mating pheromone signal transduction pathway; YHR005C GPA1 component of pheromone response pathway common to both a and alpha cells Module genes YCL065W None YKL177W None Silenced copy of ALPHA2, encoding a homeobox-domain containing protein that associates with Mcm1p in haploid cells YCL067C HMLALPHA2 to repress a-specific gene expression and interacts with A1p in diploid cells to repress haploid-specific gene expression Transcriptional