Identifying Therapeutic Vulnerabilities in Cancers Driven by the Oncogenic Transcription Factor STAT3
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
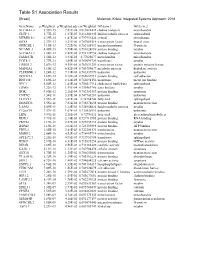
Table S1 Association Results
GeneName p.Weighted p.Weighted.adj cor.Weighted GO.term.1 GO.term.2 SLC44A1.2 3.55E-15 7.81E-08 0.819822422 choline transport mitochondria GLTP.1 5.77E-15 1.27E-07 0.816302445 lipid metabolic process sphingolipid MTMR10.1 6.39E-14 1.41E-06 0.797951424 cytosol phosphatase SOX8 2.37E-13 5.21E-06 0.787085514 transcription factor neural crest GPRC5B.1 4.19E-13 9.22E-06 0.782154937 integral membrane G-protein NCAM1.3 4.45E-13 9.79E-06 0.781624896 protein binding myelin SLC44A1.1 1.28E-12 2.82E-05 0.772132954 choline transport mitochondria FAM107B 1.56E-12 3.43E-05 0.77025677 mitochondria mitochondria UGT8.1 1.77E-12 3.89E-05 0.769099729 transferase myelin ERBB3.2 2.07E-12 4.55E-05 0.767631259 transcription factor protein tyrosine kinase MAN2A1 3.10E-12 6.82E-05 0.763759677 metabolic process hydrolase activity PLEKHH1.1 3.24E-12 7.13E-05 0.763337879 unknown unknown DOCK5.3 3.69E-12 8.12E-05 0.762087913 protein binding cell adhesion RNF130 3.69E-12 8.12E-05 0.762094156 membrane metal ion binding NPC1 6.50E-12 1.43E-04 0.756517114 cholesterol trafficking sphingolipid ERMN 7.22E-12 1.59E-04 0.755469786 actin binding myelin BOK 9.80E-12 2.16E-04 0.752383357 protein binding apoptosis CNTN2 1.54E-11 3.39E-04 0.747743281 unknown unknown ELOVL1 1.55E-11 3.41E-04 0.74764744 fatty acid sphingolipid DBNDD2 3.55E-11 7.81E-04 0.738878658 protein binding neuron projection LASS2 5.09E-11 1.12E-03 0.734954024 lipid metabolic process myelin C12orf34 7.57E-11 1.67E-03 0.730528911 unknown unknown LIPA 9.59E-11 2.11E-03 0.72786111 fatty acid glycerolipid metabolic -

Karyopherin Alpha Proteins Regulate Oligodendrocyte Differentiation
RESEARCH ARTICLE Karyopherin Alpha Proteins Regulate Oligodendrocyte Differentiation Benjamin M. Laitman1,2,3*, John N. Mariani1,2,3, Chi Zhang1,2,3, Setsu Sawai1,2,3, Gareth R. John1,2,3 1 Friedman Brain Institute, New York, New York, United States of America, 2 Corinne Goldsmith Dickinson Center for Multiple Sclerosis, New York, New York, United States of America, 3 Neurology, Icahn School of Medicine at Mount Sinai, New York, NY, New York, United States of America * [email protected] a1111111111 a1111111111 a1111111111 a1111111111 Abstract a1111111111 Proper regulation of the coordinated transcriptional program that drives oligodendrocyte (OL) differentiation is essential for central nervous system myelin formation and repair. Nuclear import, mediated in part by a group of karyopherin alpha (Kpna) proteins, regulates transcription factor access to the genome. Understanding how canonical nuclear import OPEN ACCESS functions to control genomic access in OL differentiation may aid in the creation of novel Citation: Laitman BM, Mariani JN, Zhang C, Sawai therapeutics to stimulate myelination and remyelination. Here, we show that members of S, John GR (2017) Karyopherin Alpha Proteins the Kpna family regulate OL differentiation, and may play distinct roles downstream of differ- Regulate Oligodendrocyte Differentiation. PLoS ONE 12(1): e0170477. doi:10.1371/journal. ent pro-myelinating stimuli. Multiple family members are expressed in OLs, and their phar- pone.0170477 macologic inactivation dose-dependently decreases the rate of differentiation. Additionally, Editor: Fernando de Castro, Instituto Cajal-CSIC, upon differentiation, the three major Kpna subtypes (P/α2, Q/α3, S/α1) display differential SPAIN responses to the pro-myelinating cues T3 and CNTF. -

Environmental Influences on Endothelial Gene Expression
ENDOTHELIAL CELL GENE EXPRESSION John Matthew Jeff Herbert Supervisors: Prof. Roy Bicknell and Dr. Victoria Heath PhD thesis University of Birmingham August 2012 University of Birmingham Research Archive e-theses repository This unpublished thesis/dissertation is copyright of the author and/or third parties. The intellectual property rights of the author or third parties in respect of this work are as defined by The Copyright Designs and Patents Act 1988 or as modified by any successor legislation. Any use made of information contained in this thesis/dissertation must be in accordance with that legislation and must be properly acknowledged. Further distribution or reproduction in any format is prohibited without the permission of the copyright holder. ABSTRACT Tumour angiogenesis is a vital process in the pathology of tumour development and metastasis. Targeting markers of tumour endothelium provide a means of targeted destruction of a tumours oxygen and nutrient supply via destruction of tumour vasculature, which in turn ultimately leads to beneficial consequences to patients. Although current anti -angiogenic and vascular targeting strategies help patients, more potently in combination with chemo therapy, there is still a need for more tumour endothelial marker discoveries as current treatments have cardiovascular and other side effects. For the first time, the analyses of in-vivo biotinylation of an embryonic system is performed to obtain putative vascular targets. Also for the first time, deep sequencing is applied to freshly isolated tumour and normal endothelial cells from lung, colon and bladder tissues for the identification of pan-vascular-targets. Integration of the proteomic, deep sequencing, public cDNA libraries and microarrays, delivers 5,892 putative vascular targets to the science community. -

Paraganglioma (PGL) Tumors in Patients with Succinate Dehydrogenase-Related PCC–PGL Syndromes: a Clinicopathological and Molecular Analysis
T G Papathomas and others Non-PCC/PGL tumors in the SDH 170:1 1–12 Clinical Study deficiency Non-pheochromocytoma (PCC)/paraganglioma (PGL) tumors in patients with succinate dehydrogenase-related PCC–PGL syndromes: a clinicopathological and molecular analysis Thomas G Papathomas1, Jose Gaal1, Eleonora P M Corssmit2, Lindsey Oudijk1, Esther Korpershoek1, Ketil Heimdal3, Jean-Pierre Bayley4, Hans Morreau5, Marieke van Dooren6, Konstantinos Papaspyrou7, Thomas Schreiner8, Torsten Hansen9, Per Arne Andresen10, David F Restuccia1, Ingrid van Kessel6, Geert J L H van Leenders1, Johan M Kros1, Leendert H J Looijenga1, Leo J Hofland11, Wolf Mann7, Francien H van Nederveen12, Ozgur Mete13,14, Sylvia L Asa13,14, Ronald R de Krijger1,15 and Winand N M Dinjens1 1Department of Pathology, Josephine Nefkens Institute, Erasmus MC, University Medical Center, PO Box 2040, 3000 CA Rotterdam, The Netherlands, 2Department of Endocrinology, Leiden University Medical Center, Leiden,The Netherlands, 3Section for Clinical Genetics, Department of Medical Genetics, Oslo University Hospital, Oslo, Norway, 4Department of Human and Clinical Genetics, Leiden University Medical Center, Leiden, The Netherlands, 5Department of Pathology, Leiden University Medical Center, Leiden, The Netherlands, 6Department of Clinical Genetics, Erasmus MC, University Medical Center, Rotterdam, The Netherlands, 7Department of Otorhinolaryngology, Head and Neck Surgery, University Medical Center of the Johannes Gutenberg University Mainz, Mainz, Germany, 8Section for Specialized Endocrinology, -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Regulation of Neuronal Gene Expression and Survival by Basal NMDA Receptor Activity: a Role for Histone Deacetylase 4
The Journal of Neuroscience, November 12, 2014 • 34(46):15327–15339 • 15327 Cellular/Molecular Regulation of Neuronal Gene Expression and Survival by Basal NMDA Receptor Activity: A Role for Histone Deacetylase 4 Yelin Chen,1 Yuanyuan Wang,1 Zora Modrusan,3 Morgan Sheng,1 and Joshua S. Kaminker1,2 Departments of 1Neuroscience, 2Bioinformatics and Computational Biology, and 3Molecular Biology, Genentech Inc., South San Francisco, California 94080 Neuronal gene expression is modulated by activity via calcium-permeable receptors such as NMDA receptors (NMDARs). While gene expression changes downstream of evoked NMDAR activity have been well studied, much less is known about gene expression changes that occur under conditions of basal neuronal activity. In mouse dissociated hippocampal neuronal cultures, we found that a broad NMDAR antagonist, AP5, induced robust gene expression changes under basal activity, but subtype-specific antagonists did not. While some of the gene expression changes are also known to be downstream of stimulated NMDAR activity, others appear specific to basal NMDARactivity.ThegenesalteredbyAP5treatmentofbasalcultureswereenrichedforpathwaysrelatedtoclassIIahistonedeacetylases (HDACs), apoptosis, and synapse-related signaling. Specifically, AP5 altered the expression of all three class IIa HDACs that are highly expressed in the brain, HDAC4, HDAC5, and HDAC9, and also induced nuclear accumulation of HDAC4. HDAC4 knockdown abolished a subset of the gene expression changes induced by AP5, and led to neuronal death under -

Cellular and Molecular Signatures in the Disease Tissue of Early
Cellular and Molecular Signatures in the Disease Tissue of Early Rheumatoid Arthritis Stratify Clinical Response to csDMARD-Therapy and Predict Radiographic Progression Frances Humby1,* Myles Lewis1,* Nandhini Ramamoorthi2, Jason Hackney3, Michael Barnes1, Michele Bombardieri1, Francesca Setiadi2, Stephen Kelly1, Fabiola Bene1, Maria di Cicco1, Sudeh Riahi1, Vidalba Rocher-Ros1, Nora Ng1, Ilias Lazorou1, Rebecca E. Hands1, Desiree van der Heijde4, Robert Landewé5, Annette van der Helm-van Mil4, Alberto Cauli6, Iain B. McInnes7, Christopher D. Buckley8, Ernest Choy9, Peter Taylor10, Michael J. Townsend2 & Costantino Pitzalis1 1Centre for Experimental Medicine and Rheumatology, William Harvey Research Institute, Barts and The London School of Medicine and Dentistry, Queen Mary University of London, Charterhouse Square, London EC1M 6BQ, UK. Departments of 2Biomarker Discovery OMNI, 3Bioinformatics and Computational Biology, Genentech Research and Early Development, South San Francisco, California 94080 USA 4Department of Rheumatology, Leiden University Medical Center, The Netherlands 5Department of Clinical Immunology & Rheumatology, Amsterdam Rheumatology & Immunology Center, Amsterdam, The Netherlands 6Rheumatology Unit, Department of Medical Sciences, Policlinico of the University of Cagliari, Cagliari, Italy 7Institute of Infection, Immunity and Inflammation, University of Glasgow, Glasgow G12 8TA, UK 8Rheumatology Research Group, Institute of Inflammation and Ageing (IIA), University of Birmingham, Birmingham B15 2WB, UK 9Institute of -

Noelia Díaz Blanco
Effects of environmental factors on the gonadal transcriptome of European sea bass (Dicentrarchus labrax), juvenile growth and sex ratios Noelia Díaz Blanco Ph.D. thesis 2014 Submitted in partial fulfillment of the requirements for the Ph.D. degree from the Universitat Pompeu Fabra (UPF). This work has been carried out at the Group of Biology of Reproduction (GBR), at the Department of Renewable Marine Resources of the Institute of Marine Sciences (ICM-CSIC). Thesis supervisor: Dr. Francesc Piferrer Professor d’Investigació Institut de Ciències del Mar (ICM-CSIC) i ii A mis padres A Xavi iii iv Acknowledgements This thesis has been made possible by the support of many people who in one way or another, many times unknowingly, gave me the strength to overcome this "long and winding road". First of all, I would like to thank my supervisor, Dr. Francesc Piferrer, for his patience, guidance and wise advice throughout all this Ph.D. experience. But above all, for the trust he placed on me almost seven years ago when he offered me the opportunity to be part of his team. Thanks also for teaching me how to question always everything, for sharing with me your enthusiasm for science and for giving me the opportunity of learning from you by participating in many projects, collaborations and scientific meetings. I am also thankful to my colleagues (former and present Group of Biology of Reproduction members) for your support and encouragement throughout this journey. To the “exGBRs”, thanks for helping me with my first steps into this world. Working as an undergrad with you Dr. -

(KPNA7), a Divergent Member of the Importin a Family of Nuclear Import
Kelley et al. BMC Cell Biology 2010, 11:63 http://www.biomedcentral.com/1471-2121/11/63 RESEARCH ARTICLE Open Access Karyopherin a7 (KPNA7), a divergent member of the importin a family of nuclear import receptors Joshua B Kelley1, Ashley M Talley1, Adam Spencer1, Daniel Gioeli2, Bryce M Paschal1,3* Abstract Background: Classical nuclear localization signal (NLS) dependent nuclear import is carried out by a heterodimer of importin a and importin b. NLS cargo is recognized by importin a, which is bound by importin b. Importin b mediates translocation of the complex through the central channel of the nuclear pore, and upon reaching the nucleus, RanGTP binding to importin b triggers disassembly of the complex. To date, six importin a family members, encoded by separate genes, have been described in humans. Results: We sequenced and characterized a seventh member of the importin a family of transport factors, karyopherin a 7 (KPNA7), which is most closely related to KPNA2. The domain of KPNA7 that binds Importin b (IBB) is divergent, and shows stronger binding to importin b than the IBB domains from of other importin a family members. With regard to NLS recognition, KPNA7 binds to the retinoblastoma (RB) NLS to a similar degree as KPNA2, but it fails to bind the SV40-NLS and the human nucleoplasmin (NPM) NLS. KPNA7 shows a predominantly nuclear distribution under steady state conditions, which contrasts with KPNA2 which is primarily cytoplasmic. Conclusion: KPNA7 is a novel importin a family member in humans that belongs to the importin a2 subfamily. KPNA7 shows different subcellular localization and NLS binding characteristics compared to other members of the importin a family. -

WO 2019/079361 Al 25 April 2019 (25.04.2019) W 1P O PCT
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization I International Bureau (10) International Publication Number (43) International Publication Date WO 2019/079361 Al 25 April 2019 (25.04.2019) W 1P O PCT (51) International Patent Classification: CA, CH, CL, CN, CO, CR, CU, CZ, DE, DJ, DK, DM, DO, C12Q 1/68 (2018.01) A61P 31/18 (2006.01) DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, C12Q 1/70 (2006.01) HR, HU, ID, IL, IN, IR, IS, JO, JP, KE, KG, KH, KN, KP, KR, KW, KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, (21) International Application Number: MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, PCT/US2018/056167 OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, (22) International Filing Date: SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, 16 October 2018 (16. 10.2018) TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (25) Filing Language: English (84) Designated States (unless otherwise indicated, for every kind of regional protection available): ARIPO (BW, GH, (26) Publication Language: English GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, TZ, (30) Priority Data: UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, TJ, 62/573,025 16 October 2017 (16. 10.2017) US TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, DK, EE, ES, FI, FR, GB, GR, HR, HU, ΓΕ , IS, IT, LT, LU, LV, (71) Applicant: MASSACHUSETTS INSTITUTE OF MC, MK, MT, NL, NO, PL, PT, RO, RS, SE, SI, SK, SM, TECHNOLOGY [US/US]; 77 Massachusetts Avenue, TR), OAPI (BF, BJ, CF, CG, CI, CM, GA, GN, GQ, GW, Cambridge, Massachusetts 02139 (US). -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Supplementary Materials
Supplementary materials Supplementary Table S1: MGNC compound library Ingredien Molecule Caco- Mol ID MW AlogP OB (%) BBB DL FASA- HL t Name Name 2 shengdi MOL012254 campesterol 400.8 7.63 37.58 1.34 0.98 0.7 0.21 20.2 shengdi MOL000519 coniferin 314.4 3.16 31.11 0.42 -0.2 0.3 0.27 74.6 beta- shengdi MOL000359 414.8 8.08 36.91 1.32 0.99 0.8 0.23 20.2 sitosterol pachymic shengdi MOL000289 528.9 6.54 33.63 0.1 -0.6 0.8 0 9.27 acid Poricoic acid shengdi MOL000291 484.7 5.64 30.52 -0.08 -0.9 0.8 0 8.67 B Chrysanthem shengdi MOL004492 585 8.24 38.72 0.51 -1 0.6 0.3 17.5 axanthin 20- shengdi MOL011455 Hexadecano 418.6 1.91 32.7 -0.24 -0.4 0.7 0.29 104 ylingenol huanglian MOL001454 berberine 336.4 3.45 36.86 1.24 0.57 0.8 0.19 6.57 huanglian MOL013352 Obacunone 454.6 2.68 43.29 0.01 -0.4 0.8 0.31 -13 huanglian MOL002894 berberrubine 322.4 3.2 35.74 1.07 0.17 0.7 0.24 6.46 huanglian MOL002897 epiberberine 336.4 3.45 43.09 1.17 0.4 0.8 0.19 6.1 huanglian MOL002903 (R)-Canadine 339.4 3.4 55.37 1.04 0.57 0.8 0.2 6.41 huanglian MOL002904 Berlambine 351.4 2.49 36.68 0.97 0.17 0.8 0.28 7.33 Corchorosid huanglian MOL002907 404.6 1.34 105 -0.91 -1.3 0.8 0.29 6.68 e A_qt Magnogrand huanglian MOL000622 266.4 1.18 63.71 0.02 -0.2 0.2 0.3 3.17 iolide huanglian MOL000762 Palmidin A 510.5 4.52 35.36 -0.38 -1.5 0.7 0.39 33.2 huanglian MOL000785 palmatine 352.4 3.65 64.6 1.33 0.37 0.7 0.13 2.25 huanglian MOL000098 quercetin 302.3 1.5 46.43 0.05 -0.8 0.3 0.38 14.4 huanglian MOL001458 coptisine 320.3 3.25 30.67 1.21 0.32 0.9 0.26 9.33 huanglian MOL002668 Worenine