Synthesis, Structural Characterization and Chromatographic Evaluation of Silica Hydride-Based Perfluorinated Stationary Phase
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Allyson M. Buytendyk
DISCOVERING THE ELECTRONIC PROPERTIES OF METAL HYDRIDES, METAL OXIDES AND ORGANIC MOLECULES USING ANION PHOTOELECTRON SPECTROSCOPY by Allyson M. Buytendyk A dissertation submitted to Johns Hopkins University in conformity with the requirements for the degree of Doctor of Philosophy Baltimore, Maryland September, 2015 © 2015 Allyson M. Buytendyk All Rights Reserved ABSTRACT Negatively charged molecular ions were studied in the gas phase using anion photoelectron spectroscopy. By coupling theory with the experimentally measured electronic structure, the geometries of the neutral and anion complexes could be predicted. The experiments were conducted using a one-of-a-kind time- of-flight mass spectrometer coupled with a pulsed negative ion photoelectron spectrometer. The molecules studied include metal oxides, metal hydrides, aromatic heterocylic organic compounds, and proton-coupled organic acids. Metal oxides serve as catalysts in reactions from many scientific fields and understanding the catalysis process at the molecular level could help improve reaction efficiencies - - (Chapter 1). The experimental investigation of the super-alkali anions, Li3O and Na3O , revealed both photodetachment and photoionization occur due to the low ionization potential of both neutral molecules. Additionally, HfO- and ZrO- were studied, and although both Hf and Zr have very similar atomic properties, their oxides differ greatly where ZrO- has a much lower electron affinity than HfO-. In the pursuit of using hydrogen as an environmentally friendly fuel alternative, a practical method for storing hydrogen is necessary and metal hydrides are thought to be the answer (Chapter 2). Studies yielding structural and electronic information about the hydrogen - - bonding/interacting in the complex, such as in MgH and AlH4 , are vital to constructing a practical hydrogen storage device. -

General Introduction to the Chemistry of Dyes
GENERAL INTRODUCTION TO THE CHEMISTRY OF DYES 1. Principles of Colour Chemistry 1.1 Basis for colour Unlike most organic compounds, dyes possess colour because they 1) absorb light in the visible spectrum (400–700 nm), 2) have at least one chromophore (colour-bearing group), 3) have a conjugated system, i.e. a structure with alternating double and single bonds, and 4) exhibit resonance of electrons, which is a stabilizing force in organic compounds (Abrahart, 1977). When any one of these features is lacking from the molecular structure the colour is lost. In addition to chromophores, most dyes also contain groups known as auxochromes (colour helpers), examples of which are carboxylic acid, sulfonic acid, amino, and hydroxyl groups. While these are not responsible for colour, their presence can shift the colour of a colourant and they are most often used to influence dye solubility. Figure 1 shows the relationships between wavelength of visible and colour absorbed/observed. Other factors contributing to colour are illustrated in Figures 2–4. Fig. 1. Wavelength of light absorption versus colour in organic dyes Wavelength Absorbed (nm) Colour Absorbed Colour Observed 400–435 Violet Yellow-Green 435–480 Blue Yellow 480–490 Green-Blue Orange 490–500 Blue-Green Red 500–560 Green Purple 560–580 Yellow-Green Violet 580–595 Yellow Blue 595–605 Orange Green-Blue 605–700 Red Blue-Green –55– 56 IARC MONOGRAPHS VOLUME 99 Fig. 2. Examples of chromophoric groups present in organic dyes O O N N N N O N N O H N Anthraquinone H N Nitro Azo N N Ar C N N C Ar Methine Phthalocyanine Triarylmethane Fig. -

Identification and Distribution of Dietary Precursors of The
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Vision Research 39 (1999) 219–229 Identification and distribution of dietary precursors of the Drosophila visual pigment chromophore: analysis of carotenoids in wild type and ninaD mutants by HPLC David R. Giovannucci *, Robert S. Stephenson Department of Biological Sciences, Wayne State Uni6ersity, Detroit, MI 48202, USA Received 3 July 1997; received in revised form 10 November 1997 Abstract A dietary source of retinoid or carotenoid has been shown to be necessary for the biosynthesis of functional visual pigment in flies. In the present study, the larvae or adults of Drosophila melanogaster were administered specific carotenoid-containing diets and high performance liquid chromatography was used to identify and quantify the carotenoids in extracts of wild type and ninaD visual mutant flies. When b-carotene was fed to larvae, wild type flies were shown to hydroxylate this molecule and to accumulate zeaxanthin and a small amount of b-cryptoxanthin. Zeaxanthin content was found to increase throughout development and was a major carotenoid peak detected in the adult fly. Carotenoids were twice as effective at mediating zeaxanthin accumulation when provided to larvae versus adults. In the ninaD mutant, zeaxanthin content was shown to be specifically and significantly altered compared to wild type, and was ineffective at mediating visual pigment synthesis when provided to both larval and adult mutant flies. It is proposed that zeaxanthin is the larval storage form for subsequent visual pigment chromophore biosynthesis during pupation, that zeaxanthin or b-crytoxanthin is the immediate precursor for light-independent chromophore synthesis in the adult, and that the ninaD mutant is defective in this pathway. -

Determination of Formic Acid in Acetic Acid for Industrial Use by Agilent 7820A GC
Determination of Formic Acid in Acetic Acid for Industrial Use by Agilent 7820A GC Application Brief Wenmin Liu, Chunxiao Wang HPI With rising prices of crude oil and a future shortage of oil and gas resources, people Highlights are relying on the development of the coal chemical industry. • The Agilent 7820A GC coupled with Acetic acid is an important intermediate in coal chemical synthesis. It is used in the a µTCD provides a simple method production of polyethylene, cellulose acetate, and polyvinyl, as well as synthetic for analysis of formic acid in acetic fibres and fabrics. The production of acetic acid will remain high over the next three acid. years. In China, it is estimated that the production capacity of alcohol-to-acetic acid would be 730,000 tons per year in 2010. • ALS and EPC ensure good repeata- biltiy and ease of use which makes The purity of acetic acid determinates the quality of the final synthetic products. the 7820GC appropriate for routine Formic acid is one of the main impurities in acetic acid. Many analytical methods for analysis in QA/QC labs. the analysis of formic acid in acetic acid have been developed using gas chromatog- • Using a capillary column as the ana- raphy. For example, in the GB/T 1628.5-2000 method, packed column and manual lytical column ensures better sepa- sample injection is used with poor separation and repeatability which impacts the ration of formic acid in acetic acid quantification of formic acid. compared to the China GB method. In this application brief, a new analytical method was developed on a new Agilent GC platform, the Agilent 7820A GC System. -

Ba3(P1−Xmnxo4)2 : Blue/Green Inorganic Materials Based on Tetrahedral Mn(V)
Bull. Mater. Sci., Vol. 34, No. 6, October 2011, pp. 1257–1262. c Indian Academy of Sciences. Ba3(P1−xMnxO4)2 : Blue/green inorganic materials based on tetrahedral Mn(V) SOURAV LAHA, ROHIT SHARMA, S V BHAT†, MLPREDDY‡, J GOPALAKRISHNAN∗ and S NATARAJAN Solid State and Structural Chemistry Unit, †Department of Physics, Indian Institute of Science, Bangalore 560 012, India ‡Chemical Science and Technology Division, National Institute for Interdisciplinary Science and Technology (NIIST), Thiruvananthapuram 695 019, India MS received 11 May 2011 ) 3− 2 Abstract. We describe a blue/green inorganic material, Ba3(P1−xMnxO4 2 (I) based on tetrahedral MnO4 :3d chromophore. The solid solutions (I) which are sky-blue and turquoise-blue for x ≤ 0·25 and dark green for x ≥ 0·50, 3− are readily synthesized in air from commonly available starting materials, stabilizing the MnO4 chromophore in an isostructural phosphate host. We suggest that the covalency/ionicity of P–O/Mn–O bonds in the solid solutions tunes the crystal field strength around Mn(V) such that a blue colour results for materials with small values of x. The material could serve as a nontoxic blue/green inorganic pigment. Keywords. Blue/green inorganic material; barium phosphate/manganate(V); tetrahedral manganate(V); blue/green chromophore; ligand field tuning of colour. 1. Introduction field transitions within an unusual five coordinated trigo- nal bipyramidal Mn(III). A turquoise-blue solid based on Inorganic solids displaying bright colours are important Li1·33Ti1·66O4 spinel oxide wherein the colour arises from as pigment materials which find a wide range of appli- an intervalence charge-transfer between Ti3+ and Ti4+ has cations in paints, inks, plastics, rubbers, ceramics, ena- also been described recently (Fernández-Osorio et al 2011). -
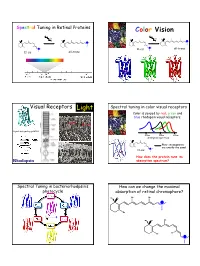
Color Vision Light
Spectral Tuning in Retinal Proteins Color Vision hν hν N N H H H H N all-trans N 11-cis 11-cis all-trans Visual Receptors Light Spectral tuning in color visual receptors Color is sensed by red, green and blue rhodopsin visual receptors. Rod Cone G-protein signaling pathway 400nm 500nm 600nm absorption spectrum Their chromophores H are exactly the same! 11-cis N How does the protein tune its Rhodopsin absorption spectrum? Spectral Tuning in bacteriorhodpsin’s How can we change the maximal photocycle absorption of retinal chromophore? Me Me Me Me N H Me Me Me Me N H Me Me Me Me Me Me Me H N 1 Excitation energy determines the Electronic Absorption maximal absorption Me Me Me Me π π* N - H S1 Me Absorption of light in the UV-VIS region of the S0 spectrum is due to Response excitation of electrons to higher energy levels. Me Me Me 13 7911 15 N H Me Me π-π* excitation in polyenes π-π* excitation in polyenes π∗ π∗ π∗ π∗ ∆E π∗ E photon E π π π π π Ground state (S0) Excited state (S1) ∆E (excitation energy, band gap) = hν = hc/λ blue-shift red-shift π-π* excitation in polyenes Tuning the length of the conjugated backbone β-carotene O O Vitamin A2 (retinal II) Vitamin A1 (retinal I) Longer wavelength Short wavelength 2 Retinal I Retinal II OPSIN SHIFT: how protein tunes the absorption maximum of its chromophore. Maximal absorption of protonated retinal Schiff base in: Water/methanol solution: 440 nm bR: 568 nm rod Rh: 500 nm red receptor: 560 nm green receptor: 530 nm blue receptor: 426 nm Salmon: different retinals in different stages of life Electrostatics and opsin shift Electrostatics and opsin shift S S positive charge 2 positive charge 2 S2 S2 Me Me Me Me Me Me S1 S1 S1 S1 + + + + N N H H Me O S0 Me O S0 Me O Me O C C Asp (Glu) Asp (Glu) S0 S0 no protein in protein no protein in protein counterion counterion • The counterion stabilizes the positive charge of the chromophore. -

Basis for Listing Hazardous Waste
NEBRASKA ADMINISTRATIVE CODE Title 128 - Department of Environmental Quality Appendix II - BASIS FOR LISTING HAZARDOUS WASTE EPA Hazardous Hazardous Constituents For Which Listed Waste No. F001 Tetrachloroethylene; methylene chloride; trichloroethylene; 1,1,1-trichloroethane; carbon tetrachloride; chlorinated fluorocarbons. F002 Tetrachloroethylene; methylene chloride; trichloroethylene; 1,1,1-trichloroethane; 1,1,2-trichloroethane; chlorobenzene; 1,1,2-trichloro-1,2,2-trichfluoroethane; ortho- dichlorobenzene; trichlorofluoromethane. F003 N.A. F004 Cresols and cresylic acid, nitrobenzene. F005 Toluene, methyl ethyl ketone, carbon disulfide, isobutanol, pyridine, 2-ethoxyethanol, benzene, 2-nitropropane. F006 Cadmium, hexavalent chromium, nickel, cyanide (complexed). F007 Cyanide (salts). F008 Cyanide (salts). F009 Cyanide (salts). F010 Cyanide (salts). F011 Cyanide (salts). F012 Cyanide (complexed). F019 Hexavalent chromium, cyanide (complexed). F020 Tetra- and pentachlorodibenzo-p-dioxins; tetra- and pentachlorodibenzofurans; tri- and tetrachlorophenols and their chlorophenoxy derivative acids, esters, ethers, amine and other salts. Effective Date: 01/03/07 II-1 Title 128 Appendix II EPA Hazardous Hazardous Constituents For Which Listed Waste No. F021 Penta- and hexachlorodibenzo-p-dioxins; penta- and hexachlorodibenzofurans; pentachlorophenol and its derivatives. F022 Tetra-, penta-, and hexachlorodibenzo-p-dioxins; tetra-, penta-, and hexachlorodibenzofurans. F023 Tetra-, and pentachlorodibenzo-p-dioxins; tetra- and pentachlorodibenzofurans; -

Radical-Radical Reactions, Pyrene Nucleation, and Incipient Soot
Available online at www.sciencedirect.com Proceedings of the Combustion Institute 36 (2017) 799–806 www.elsevier.com/locate/proci Radical–radical reactions, pyrene nucleation, and incipient soot formation in combustion a b b K. Olof Johansson , Tyler Dillstrom , Paolo Elvati , a a c ,d Matthew F. Campbell , Paul E. Schrader , Denisia M. Popolan-Vaida , c c b ,e Nicole K. Richards-Henderson , Kevin R. Wilson , Angela Violi , a , ∗ Hope A. Michelsen a Combustion Research Facility, Sandia National Laboratories, P. O. Box 969, MS 9055, Livermore, CA 94551, USA b Department of Mechanical Engineering, University of Michigan, Ann Arbor, MI 48109, USA c Chemical Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA d Department of Chemistry, University of California, Berkeley, CA 94720, USA e Departments of Chemical Engineering, Biomedical Engineering, Macromolecular Science and Engineering, Biophysics Program, University of Michigan, Ann Arbor, MI 48109, USA Received 3 December 2015; accepted 31 July 2016 Available online 12 October 2016 Abstract We present a combined experimental and probabilistic simulation study of soot-precursor. The experi- ments were conducted using aerosol mass spectrometry coupled with tunable vacuum ultraviolet radiation from the Advanced Light Source at Lawrence Berkeley National Laboratory. Mass spectra and photoion- ization efficiency (PIE) curves of soot precursor species were measured at different heights in a premixed flat flame and in a counter-flow diffusion flame fueled by ethylene and oxygen. The PIE curves at the pyrene mass from these flames were compared with reference PIE scans recorded for pyrene. The results demonstrate that other C 16 H 10 isomers than pyrene are major components among species condensed onto incipient soot in this study, which is in agreement with the simulations. -

Temperature-Induced Oligomerization of Polycyclic Aromatic Hydrocarbons
www.nature.com/scientificreports OPEN Temperature-induced oligomerization of polycyclic aromatic hydrocarbons at ambient Received: 7 June 2017 Accepted: 10 July 2017 and high pressures Published: xx xx xxxx Artem D. Chanyshev 1,2, Konstantin D. Litasov1,2, Yoshihiro Furukawa3, Konstantin A. Kokh1,2 & Anton F. Shatskiy1,2 Temperature-induced oligomerization of polycyclic aromatic hydrocarbons (PAHs) was found at 500–773 K and ambient and high (3.5 GPa) pressures. The most intensive oligomerization at 1 bar and 3.5 GPa occurs at 740–823 K. PAH carbonization at high pressure is the fnal stage of oligomerization and occurs as a result of sequential oligomerization and polymerization of the starting material, caused by overlapping of π-orbitals, a decrease of intermolecular distances, and fnally the dehydrogenation and polycondensation of benzene rings. Being important for building blocks of life, PAHs and their oligomers can be formed in the interior of the terrestrial planets with radii less than 2270 km. High-pressure transformations of polycyclic aromatic hydrocarbons (PAHs) and benzene become extremely important due to wide applications for example in graphene- and graphene-based nanotechnology1–3, synthesis of organic superconductors4, 5, petroleum geoscience, origin of organic molecules in Universe and origin of life. In particular, PAHs were found in many space objects: meteorites6–8, cometary comae9, interstellar clouds and planetary nebulas10–12. Although the prevalent hypothesis for the formation of these PAHs is irradiation-driven polymerization of smaller hydrocarbons13, alternative explanation could be shock fragmentation of carbonaceous solid material11. PAH-bearing carbonaceous material could contribute to the delivery of extraterrestrial organic materials to the prebiotic Earth during the period of heavy bombardment of the inner Solar System from 4.5 to 3.8 Ga ago14–16. -

Ester Synthesis Lab (Student Handout)
Name: ________________________ Lab Partner: ____________________ Date: __________________________ Class Period: ____________________ Ester Synthesis Lab (Student Handout) Lab Report Components: The following must be included in your lab book in order to receive full credit. 1. Purpose 2. Hypothesis 3. Procedure 4. Observation/Data Table 5. Results 6. Mechanism (In class) 7. Conclusion Introduction The compounds you will be making are also naturally occurring compounds; the chemical structure of these compounds is already known from other investigations. Esters are organic molecules of the general form: where R1 and R2 are any carbon chain. Esters are unique in that they often have strong, pleasant odors. As such, they are often used in fragrances, and many artificial flavorings are in fact esters. Esters are produced by the reaction between alcohols and carboxylic acids. For example, reacting ethanol with acetic acid to give ethyl acetate is shown below. + → + In the case of ethyl acetate, R1 is a CH3 group and R2 is a CH3CH2 group. Naming esters systematically requires naming the functional groups on both sides of the bridging oxygen. In the example above, the right side of the ester as shown is a CH3CH2 1 group, or ethyl group. The left side is CH3C=O, or acetate. The name of the ester is therefore ethyl acetate. Deriving the names of the side from the carboxylic acid merely requires replacing the suffix –ic with –ate. Materials • Alcohol • Carboxylic Acid o 1 o A o 2 o B o 3 o C o 4 Observation Parameters: • Record the combination of carboxylic acid and alcohol • Observe each reactant • Observe each product Procedure 1. -

Supplement of Investigation of Secondary Formation of Formic Acid: Urban Environment Vs
Supplement of Atmos. Chem. Phys. Discuss., 14, 24863–24914, 2014 http://www.atmos-chem-phys-discuss.net/14/24863/2014/ doi:10.5194/acpd-14-24863-2014-supplement © Author(s) 2014. CC Attribution 3.0 License. Supplement of Investigation of secondary formation of formic acid: urban environment vs. oil and gas producing region B. Yuan et al. Correspondence to: B. Yuan ([email protected]) 31 Table S1. Comparisons of measured and modeled formic acid in previous studies. Studies Location and time Model Notes Atlantic Ocean 1 MOGUNTIA Model results are 8 times lower than observations. (1996.10-11) Amazonia, Congo, Model underestimates HCOOH both in free 2 MOGUNTIA Virginia troposphere and boundary layer. Marine air, and MATCH- Measurements are underestimated by a factor of 2 or 3 those in Poisson et MPIC more, except in Amazon. al. MATCH- Measured concentrations are substantially higher than 4 Kitt Peak, US MPIC the model-calculated values. Model underestimates HCOOH compared to 5 Various sites IMPACT observations at most sites. Various sites Sources of formic acid may be up to 50% greater than 6 (MILAGRO, GEOS-Chem the estimates and the study reports evidence of a long- INTEX-B) lived missing secondary source of formic acid. Model underestimates HCOOH concentrations by up Trajectory to a factor of 2. The missing sources are considered to model with 7 North Sea (2010.3) be both primary emissions of HCOOH of MCM V3.2 & anthropogenic origin and a lack of precursor CRI emissions, e.g. isoprene. The globally source of formic acid (100-120 Tg) is 2-3 Satellite times more than that estimated from known sources. -
Biomass Oxidation to Formic Acid in Aqueous Media Using Polyoxometalate Catalysts – Boosting FA Selectivity by In-Situ Extraction
Electronic Supplementary Material (ESI) for Energy & Environmental Science. This journal is © The Royal Society of Chemistry 2015 Biomass Oxidation to Formic Acid in Aqueous Media Using Polyoxometalate Catalysts – Boosting FA Selectivity by In-situ Extraction Supplementary Information Determination of distribution coefficients A model reaction solution containing 0.91 g of the HPA-5 catalyst, 10.18 g FA and 50.0 g water was prepared. This solution was stirred with 50.91 g of the extracting solvent at 363 K for one hour and then transferred into a separation funnel. After phase separation, a sample of each phase was taken and analysed by means of 1H-NMR to determine the formic acid concentration. Extraction Solvent Screening Table S1: Solvent screening to identify a suitable extraction solvent - extraction of a simulated product solution. Distribution coefficient K Selectivity Extracting agent a b cFA,org./cFA,aqu KFA/Kwater 1-Hexanol 0.94 8.6 1-Heptanol 0.67 4.4 Butylethylether 0.49 2.7 Benzyl formate 0.46 2.6 Heptyl formate 0.42 2.2 Di-isopropylether 0.40 2.5 Di-n-butylether 0.22 1.2 Conditions: 10.18 g formic acid, 0.91 g (0.5 mmol) HPA-5 catalyst dissolved in 50.0 mL H2O, together with 50.91 g of the extracting solvent; stirred at 363 K; 1 h. a) as determined by 1H-NMR using benzene as external standard; b) ratio of the distribution coefficients of formic acid and water. Table S2: Esterification activity of formic acid with 1-hexanol and 1-heptanol, respectively.