A Sparse and Low-Rank Regression Model for Identifying The
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Integrative Genomic and Epigenomic Analyses Identified IRAK1 As a Novel Target for Chronic Inflammation-Driven Prostate Tumorigenesis
bioRxiv preprint doi: https://doi.org/10.1101/2021.06.16.447920; this version posted June 16, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Integrative genomic and epigenomic analyses identified IRAK1 as a novel target for chronic inflammation-driven prostate tumorigenesis Saheed Oluwasina Oseni1,*, Olayinka Adebayo2, Adeyinka Adebayo3, Alexander Kwakye4, Mirjana Pavlovic5, Waseem Asghar5, James Hartmann1, Gregg B. Fields6, and James Kumi-Diaka1 Affiliations 1 Department of Biological Sciences, Florida Atlantic University, Florida, USA 2 Morehouse School of Medicine, Atlanta, Georgia, USA 3 Georgia Institute of Technology, Atlanta, Georgia, USA 4 College of Medicine, Florida Atlantic University, Florida, USA 5 Department of Computer and Electrical Engineering, Florida Atlantic University, Florida, USA 6 Department of Chemistry & Biochemistry and I-HEALTH, Florida Atlantic University, Florida, USA Corresponding Author: [email protected] (S.O.O) Running Title: Chronic inflammation signaling in prostate tumorigenesis bioRxiv preprint doi: https://doi.org/10.1101/2021.06.16.447920; this version posted June 16, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Abstract The impacts of many inflammatory genes in prostate tumorigenesis remain understudied despite the increasing evidence that associates chronic inflammation with prostate cancer (PCa) initiation, progression, and therapy resistance. -

Spatially Heterogeneous Choroid Plexus Transcriptomes Encode Positional Identity and Contribute to Regional CSF Production
The Journal of Neuroscience, March 25, 2015 • 35(12):4903–4916 • 4903 Development/Plasticity/Repair Spatially Heterogeneous Choroid Plexus Transcriptomes Encode Positional Identity and Contribute to Regional CSF Production Melody P. Lun,1,3 XMatthew B. Johnson,2 Kevin G. Broadbelt,1 Momoko Watanabe,4 Young-jin Kang,4 Kevin F. Chau,1 Mark W. Springel,1 Alexandra Malesz,1 Andre´ M.M. Sousa,5 XMihovil Pletikos,5 XTais Adelita,1,6 Monica L. Calicchio,1 Yong Zhang,7 Michael J. Holtzman,7 Hart G.W. Lidov,1 XNenad Sestan,5 Hanno Steen,1 XEdwin S. Monuki,4 and Maria K. Lehtinen1 1Department of Pathology, and 2Division of Genetics, Boston Children’s Hospital, Boston, Massachusetts 02115, 3Department of Pathology and Laboratory Medicine, Boston University School of Medicine, Boston, Massachusetts 02118, 4Department of Pathology and Laboratory Medicine, University of California Irvine School of Medicine, Irvine, California 92697, 5Department of Neurobiology and Kavli Institute for Neuroscience, Yale School of Medicine, New Haven, Connecticut 06510, 6Department of Biochemistry, Federal University of Sa˜o Paulo, Sa˜o Paulo 04039, Brazil, and 7Pulmonary and Critical Care Medicine, Department of Medicine, Washington University, St Louis, Missouri 63110 A sheet of choroid plexus epithelial cells extends into each cerebral ventricle and secretes signaling factors into the CSF. To evaluate whether differences in the CSF proteome across ventricles arise, in part, from regional differences in choroid plexus gene expression, we defined the transcriptome of lateral ventricle (telencephalic) versus fourth ventricle (hindbrain) choroid plexus. We find that positional identitiesofmouse,macaque,andhumanchoroidplexiderivefromgeneexpressiondomainsthatparalleltheiraxialtissuesoforigin.We thenshowthatmolecularheterogeneitybetweentelencephalicandhindbrainchoroidplexicontributestoregion-specific,age-dependent protein secretion in vitro. -

Fog2 Is Critical for Cardiac Function and Maintenance of Coronary Vasculature in the Adult Mouse Heart
Fog2 is critical for cardiac function and maintenance of coronary vasculature in the adult mouse heart Bin Zhou, … , Sergei G. Tevosian, William T. Pu J Clin Invest. 2009;119(6):1462-1476. https://doi.org/10.1172/JCI38723. Research Article Cardiology Aberrant transcriptional regulation contributes to the pathogenesis of both congenital and adult forms of heart disease. While the transcriptional regulator friend of Gata 2 (FOG2) is known to be essential for heart morphogenesis and coronary development, its tissue-specific function has not been previously investigated. Additionally, little is known about the role of FOG2 in the adult heart. Here we used spatiotemporally regulated inactivation of Fog2 to delineate its function in both the embryonic and adult mouse heart. Early cardiomyocyte-restricted loss of Fog2 recapitulated the cardiac and coronary defects of the Fog2 germline murine knockouts. Later cardiomyocyte-restricted loss ofF og2 (Fog2MC) did not result in defects in cardiac structure or coronary vessel formation. However, Fog2MC adult mice had severely depressed ventricular function and died at 8–14 weeks. Fog2MC adult hearts displayed a paucity of coronary vessels, associated with myocardial hypoxia, increased cardiomyocyte apoptosis, and cardiac fibrosis. Induced inactivation of Fog2 in the adult mouse heart resulted in similar phenotypes, as did ablation of the FOG2 interaction with the transcription factor GATA4. Loss of the FOG2 or FOG2-GATA4 interaction altered the expression of a panel of angiogenesis-related genes. Collectively, our data indicate that FOG2 regulates adult heart function and coronary angiogenesis. Find the latest version: https://jci.me/38723/pdf Research article Fog2 is critical for cardiac function and maintenance of coronary vasculature in the adult mouse heart Bin Zhou,1,2 Qing Ma,1 Sek Won Kong,1 Yongwu Hu,1,3 Patrick H. -

Mechanism of Lncrna FEZF1-AS1 in Promoting the Occurrence and Development of Oral Squamous Cell Carcinoma Through Targeting Mir-196A
European Review for Medical and Pharmacological Sciences 2019; 23: 6505-6515 Mechanism of lncRNA FEZF1-AS1 in promoting the occurrence and development of oral squamous cell carcinoma through targeting miR-196a L. XU, T.-J. HOU, P. YANG Department of Stomatology, Liaocheng People’s Hospital, Liaocheng, China Lin Xu and Tiejun Hou contributed equally to this work Abstract. – OBJECTIVE: Previous studies have Meanwhile, the miR-196a expression was nega- demonstrated that long non-coding ribonucleic tively correlated with FEZF1-AS1. Subsequent acid (lncRNA) FEZF1-AS1 acts as a cancer-pro- Luciferase reporter gene assay confirmed that moting gene. However, no reports have investi- overexpression of miR-196a could markedly re- gated the role of FEZF1-AS1 in oral squamous cell duce the activity of Luciferase containing wild- carcinoma (OSCC) yet. Therefore, the aim of this type FEZF1-AS1 vector rather than decrease the study was to explore whether FEZF1-AS1 promot- activity of Luciferase containing mutant-type ed the expression characteristics of OSCC by tar- vector or empty vector. These findings further geting miR-196a and to further elucidate the un- indicated that FEZF1-AS1 could be targeted by derlying mechanism of FEZF1-AS1 in promoting miR-196a through this binding site. In addition, the metastasis of OSCC. recovery assay demonstrates that there was a PATIENTS AND METHODS: The expression mutual regulatory effect between FEZF1-AS1 levels of FEZF1-AS1 and miR-196a in 42 pairs of and miR-196a, jointly affecting the malignant OSCC tissues and para-carcinoma tissues were progression of OSCC. detected via quantitative Real Time-Polymerase CONCLUSIONS: The expression of lncRNA Chain Reaction (qRT-PCR). -

Proteomics Provides Insights Into the Inhibition of Chinese Hamster V79
www.nature.com/scientificreports OPEN Proteomics provides insights into the inhibition of Chinese hamster V79 cell proliferation in the deep underground environment Jifeng Liu1,2, Tengfei Ma1,2, Mingzhong Gao3, Yilin Liu4, Jun Liu1, Shichao Wang2, Yike Xie2, Ling Wang2, Juan Cheng2, Shixi Liu1*, Jian Zou1,2*, Jiang Wu2, Weimin Li2 & Heping Xie2,3,5 As resources in the shallow depths of the earth exhausted, people will spend extended periods of time in the deep underground space. However, little is known about the deep underground environment afecting the health of organisms. Hence, we established both deep underground laboratory (DUGL) and above ground laboratory (AGL) to investigate the efect of environmental factors on organisms. Six environmental parameters were monitored in the DUGL and AGL. Growth curves were recorded and tandem mass tag (TMT) proteomics analysis were performed to explore the proliferative ability and diferentially abundant proteins (DAPs) in V79 cells (a cell line widely used in biological study in DUGLs) cultured in the DUGL and AGL. Parallel Reaction Monitoring was conducted to verify the TMT results. γ ray dose rate showed the most detectable diference between the two laboratories, whereby γ ray dose rate was signifcantly lower in the DUGL compared to the AGL. V79 cell proliferation was slower in the DUGL. Quantitative proteomics detected 980 DAPs (absolute fold change ≥ 1.2, p < 0.05) between V79 cells cultured in the DUGL and AGL. Of these, 576 proteins were up-regulated and 404 proteins were down-regulated in V79 cells cultured in the DUGL. KEGG pathway analysis revealed that seven pathways (e.g. -

Targeting Non-Oncogene Addiction for Cancer Therapy
biomolecules Review Targeting Non-Oncogene Addiction for Cancer Therapy Hae Ryung Chang 1,*,†, Eunyoung Jung 1,†, Soobin Cho 1, Young-Jun Jeon 2 and Yonghwan Kim 1,* 1 Department of Biological Sciences and Research Institute of Women’s Health, Sookmyung Women’s University, Seoul 04310, Korea; [email protected] (E.J.); [email protected] (S.C.) 2 Department of Integrative Biotechnology, Sungkyunkwan University, Suwon 16419, Korea; [email protected] * Correspondence: [email protected] (H.R.C.); [email protected] (Y.K.); Tel.: +82-2-710-9552 (H.R.C.); +82-2-710-9552 (Y.K.) † These authors contributed equally. Abstract: While Next-Generation Sequencing (NGS) and technological advances have been useful in identifying genetic profiles of tumorigenesis, novel target proteins and various clinical biomarkers, cancer continues to be a major global health threat. DNA replication, DNA damage response (DDR) and repair, and cell cycle regulation continue to be essential systems in targeted cancer therapies. Although many genes involved in DDR are known to be tumor suppressor genes, cancer cells are often dependent and addicted to these genes, making them excellent therapeutic targets. In this review, genes implicated in DNA replication, DDR, DNA repair, cell cycle regulation are discussed with reference to peptide or small molecule inhibitors which may prove therapeutic in cancer patients. Additionally, the potential of utilizing novel synthetic lethal genes in these pathways is examined, providing possible new targets for future therapeutics. Specifically, we evaluate the potential of TONSL as a novel gene for targeted therapy. Although it is a scaffold protein with no known enzymatic activity, the strategy used for developing PCNA inhibitors can also be utilized to target TONSL. -

Epigenetic Mechanisms of Lncrnas Binding to Protein in Carcinogenesis
cancers Review Epigenetic Mechanisms of LncRNAs Binding to Protein in Carcinogenesis Tae-Jin Shin, Kang-Hoon Lee and Je-Yoel Cho * Department of Biochemistry, BK21 Plus and Research Institute for Veterinary Science, School of Veterinary Medicine, Seoul National University, Seoul 08826, Korea; [email protected] (T.-J.S.); [email protected] (K.-H.L.) * Correspondence: [email protected]; Tel.: +82-02-800-1268 Received: 21 September 2020; Accepted: 9 October 2020; Published: 11 October 2020 Simple Summary: The functional analysis of lncRNA, which has recently been investigated in various fields of biological research, is critical to understanding the delicate control of cells and the occurrence of diseases. The interaction between proteins and lncRNA, which has been found to be a major mechanism, has been reported to play an important role in cancer development and progress. This review thus organized the lncRNAs and related proteins involved in the cancer process, from carcinogenesis to metastasis and resistance to chemotherapy, to better understand cancer and to further develop new treatments for it. This will provide a new perspective on clinical cancer diagnosis, prognosis, and treatment. Abstract: Epigenetic dysregulation is an important feature for cancer initiation and progression. Long non-coding RNAs (lncRNAs) are transcripts that stably present as RNA forms with no translated protein and have lengths larger than 200 nucleotides. LncRNA can epigenetically regulate either oncogenes or tumor suppressor genes. Nowadays, the combined research of lncRNA plus protein analysis is gaining more attention. LncRNA controls gene expression directly by binding to transcription factors of target genes and indirectly by complexing with other proteins to bind to target proteins and cause protein degradation, reduced protein stability, or interference with the binding of other proteins. -

Functional Characterization of Alternative Splice Variants of The
Functional characterization of alternative splice variants of the Drosophila GATA transcription factor serpent containing either one or two zinc finger domains Douaa Moussalem To cite this version: Douaa Moussalem. Functional characterization of alternative splice variants of the Drosophila GATA transcription factor serpent containing either one or two zinc finger domains. Genetics. Université Paul Sabatier - Toulouse III, 2020. English. NNT : 2020TOU30138. tel-03162582 HAL Id: tel-03162582 https://tel.archives-ouvertes.fr/tel-03162582 Submitted on 8 Mar 2021 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. THÈSE En vue de l’obtention du DOCTORAT DE L’UNIVERSITÉ DE TOULOUSE Délivré par l'Université Toulouse 3 - Paul Sabatier Présentée et soutenue par Douaa MOUSSALEM Le 12 octobre 2020 Titre : Functional characterization of alternative splice variants of the Drosophila GATA transcription factor Serpent containing either one or two zinc finger domains. Ecole doctorale : BSB - Biologie, Santé, Biotechnologies Spécialité : GENETIQUE MOLECULAIRE Unité de recherche : CBD - Centre de Biologie du Développement Thèse dirigée par Marc HAENLIN et Dani OSMAN Jury M. David CRIBBS, Président Mme Annarita MICCIO, Rapporteure Mme Kyra CAMPBELL, Rapporteure M. Samir MERABET, Rapporteur M. Marc HAENLIN, Directeur de thèse M. -

Plenary and Platform Abstracts
American Society of Human Genetics 68th Annual Meeting PLENARY AND PLATFORM ABSTRACTS Abstract #'s Tuesday, October 16, 5:30-6:50 pm: 4. Featured Plenary Abstract Session I Hall C #1-#4 Wednesday, October 17, 9:00-10:00 am, Concurrent Platform Session A: 6. Variant Insights from Large Population Datasets Ballroom 20A #5-#8 7. GWAS in Combined Cancer Phenotypes Ballroom 20BC #9-#12 8. Genome-wide Epigenomics and Non-coding Variants Ballroom 20D #13-#16 9. Clonal Mosaicism in Cancer, Alzheimer's Disease, and Healthy Room 6A #17-#20 Tissue 10. Genetics of Behavioral Traits and Diseases Room 6B #21-#24 11. New Frontiers in Computational Genomics Room 6C #25-#28 12. Bone and Muscle: Identifying Causal Genes Room 6D #29-#32 13. Precision Medicine Initiatives: Outcomes and Lessons Learned Room 6E #33-#36 14. Environmental Exposures in Human Traits Room 6F #37-#40 Wednesday, October 17, 4:15-5:45 pm, Concurrent Platform Session B: 24. Variant Interpretation Practices and Resources Ballroom 20A #41-#46 25. Integrated Variant Analysis in Cancer Genomics Ballroom 20BC #47-#52 26. Gene Discovery and Functional Models of Neurological Disorders Ballroom 20D #53-#58 27. Whole Exome and Whole Genome Associations Room 6A #59-#64 28. Sequencing-based Diagnostics for Newborns and Infants Room 6B #65-#70 29. Omics Studies in Alzheimer's Disease Room 6C #71-#76 30. Cardiac, Valvular, and Vascular Disorders Room 6D #77-#82 31. Natural Selection and Human Phenotypes Room 6E #83-#88 32. Genetics of Cardiometabolic Traits Room 6F #89-#94 Wednesday, October 17, 6:00-7:00 pm, Concurrent Platform Session C: 33. -

Transcription Factors from Multiple Families Ensure Enhancer Selectivity
bioRxiv preprint doi: https://doi.org/10.1101/2020.09.04.283036; this version posted September 4, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Transcription factors from multiple families ensure enhancer selectivity and robust neuron terminal differentiation Angela Jimeno-Martín, Erick Sousa, Noemi Daroqui, Rebeca Brocal-Ruiz, Miren Maicas, Nuria Flames* Developmental Neurobiology Unit, Instituto de Biomedicina de Valencia IBV- CSIC, Valencia, 46010, Spain * Correspondence: [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.09.04.283036; this version posted September 4, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 SUMMARY 2 To search for general principles underlying neuronal regulatory programs we 3 built an RNA interference library against all transcription factors (TFs) encoded 4 in C. elegans genome and systematically screened for specification defects in 5 ten different neuron types of the monoaminergic (MA) superclass. 6 We identified over 90 TFs involved in MA specification, with at least ten different 7 TFs controlling differentiation of each individual neuron type. These TFs belong 8 predominantly to five TF families (HD, bHLH, ZF, bZIP and NHR). Next, 9 focusing on the complexity of terminal differentiation, we identified and 10 functionally characterized the dopaminergic terminal regulatory program. We 11 found that seven TFs from four different families act in a TF collective to provide 12 genetic robustness and to impose a specific gene regulatory signature enriched 13 in the regulatory regions of dopamine effector genes. -
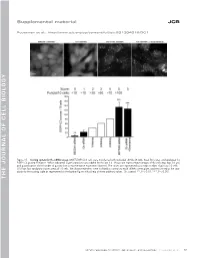
Thejournalofcellb Io Logy
Supplemental material JCB Puustinen et al., http://www.jcb.org/cgi/content/full/jcb.201304012/DC1 Figure S1. Scoring system for the siRNA screen. MCF7-EGFP-LC3 cells were transfected with indicated siRNAs (8 nM), fixed 56 h later, and analyzed for EGFP-LC3 puncta formation. When indicated, 5 µM siramesine was added for the last 3 h. Shown are representative images of the cells (top; bar, 10 µm) and quantification of the number of puncta from a representative experiment (bottom). The values are represented as a mean number of puncta/10 cells ± SDs from four randomly chosen areas of 10 cells. The shown treatments were included as controls to each siRNA screen plate, and they served as the stan- dards for the scoring scale as represented on the bottom figure with a help of three arbitrary values. Ctr, control. **, P < 0.01; ***, P < 0.001. THE JOURNAL OF CELL BIOLOGY CIP2A regulates mTORC1, cell growth, and autophagy • Puustinen et al. S1 Figure S2. A cartoon highlighting the positions of the candidate autophagy-regulating genes in the insulin receptor signaling network. The cartoon was prepared by Ingenuity Pathway Analysis. software. CP, carbohydrate phosphatase; ERK, extracellular signal–regulated kinase; FSH, follicle-stimulating hor- mone; EG, epidermal growth factor. S2 JCB Figure S3. The effect of siRNAs targeting the autophagy-regulating PP2 subunits on target gene expression, cell density, and mTORC1 pathway. (A) Total RNA was isolated from MCF7 cells treated with indicated siRNAs (20 nM) for 54 h and analyzed for target gene expression by qPCR using primers de- signed to specifically amplify the indicated PP2A-related mRNAs andACTB mRNA (internal control). -

Supplementary Materials
Supplementary materials Supplementary Table S1: MGNC compound library Ingredien Molecule Caco- Mol ID MW AlogP OB (%) BBB DL FASA- HL t Name Name 2 shengdi MOL012254 campesterol 400.8 7.63 37.58 1.34 0.98 0.7 0.21 20.2 shengdi MOL000519 coniferin 314.4 3.16 31.11 0.42 -0.2 0.3 0.27 74.6 beta- shengdi MOL000359 414.8 8.08 36.91 1.32 0.99 0.8 0.23 20.2 sitosterol pachymic shengdi MOL000289 528.9 6.54 33.63 0.1 -0.6 0.8 0 9.27 acid Poricoic acid shengdi MOL000291 484.7 5.64 30.52 -0.08 -0.9 0.8 0 8.67 B Chrysanthem shengdi MOL004492 585 8.24 38.72 0.51 -1 0.6 0.3 17.5 axanthin 20- shengdi MOL011455 Hexadecano 418.6 1.91 32.7 -0.24 -0.4 0.7 0.29 104 ylingenol huanglian MOL001454 berberine 336.4 3.45 36.86 1.24 0.57 0.8 0.19 6.57 huanglian MOL013352 Obacunone 454.6 2.68 43.29 0.01 -0.4 0.8 0.31 -13 huanglian MOL002894 berberrubine 322.4 3.2 35.74 1.07 0.17 0.7 0.24 6.46 huanglian MOL002897 epiberberine 336.4 3.45 43.09 1.17 0.4 0.8 0.19 6.1 huanglian MOL002903 (R)-Canadine 339.4 3.4 55.37 1.04 0.57 0.8 0.2 6.41 huanglian MOL002904 Berlambine 351.4 2.49 36.68 0.97 0.17 0.8 0.28 7.33 Corchorosid huanglian MOL002907 404.6 1.34 105 -0.91 -1.3 0.8 0.29 6.68 e A_qt Magnogrand huanglian MOL000622 266.4 1.18 63.71 0.02 -0.2 0.2 0.3 3.17 iolide huanglian MOL000762 Palmidin A 510.5 4.52 35.36 -0.38 -1.5 0.7 0.39 33.2 huanglian MOL000785 palmatine 352.4 3.65 64.6 1.33 0.37 0.7 0.13 2.25 huanglian MOL000098 quercetin 302.3 1.5 46.43 0.05 -0.8 0.3 0.38 14.4 huanglian MOL001458 coptisine 320.3 3.25 30.67 1.21 0.32 0.9 0.26 9.33 huanglian MOL002668 Worenine