The Determination of Sulfur As Barium Sulfate
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
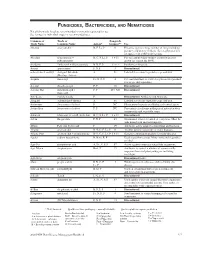
Fungicides, Bactericides, and Nematicides Not All Chemicals Listed Are Recommended Or Currently Registered for Use
FUNGICIDES, BACTERICIDES, AND NEMATICIDES Not all chemicals listed are recommended or currently registered for use. See listings for individual crops for recommended uses. Common or Trade or Fungicide Trade Name Common Name Action* Group #** Use Abound azoxystrobin B, F, Ls, P 11 Effective against a large number of fungi including powdery and downy mildews. Severe phytotoxicity on apples with a McIntosh heritage. Absolute tebuconazole + B, C, F, Ls, P 3 + 11 For rust and powdery mildew control in grasses trifloxystrobin grown for seed in the PNW. Academy fludioxonil + difenoconazole B-N, F, P 12 + 3 Postharvest fungicide. Accrue spiroxamine F, N, P 5 Discontinued. acibenzolar-S-methyl Actigard, Blockade A P1 Labeled for certain vegetable crops and fruit. (Heritage Action) Acquire metalaxyl Fs, N, P, S 4 For seed treatment to control ooymcetes in specified row crops and vegetables. Acrobat dimethomorph F, P 40 Discontinued. Acrobat MZ dimethomorph + F, P 40 + M3 Discontinued. mancozeb Acti-dione cycloheximide F Discontinued. Antibiotic and fungicide. Actigard acibenzolar-S-methyl A P1 Labeled for certain vegetable crops and fruit. Actinovate Streptomyces lydicus F NC Filamentous bacteria as a Biological control agent. Actino-Iron Streptomyces lydicus F, P NC For control of soilborne pathogens of indoor/outdoor ornamentals and vegetable crops. Adament tebuconazole + trifloxystrobin B, C, F, Ls, P 3 + 11 Discontinued. Adorn fluopicolide F, N, P 43 Ornamental label for control of oomycetes. Must be tank-mixed with another fungicide. Affirm Polyoxin D zinc salt F 19 Antibiotic active against certain fungi and bacteria. Aframe azoxystrobin B-N, C, F, Ls, P 11 Another generic fungicide for many diseases. -

Historical Perspectives on Apple Production: Fruit Tree Pest Management, Regulation and New Insecticidal Chemistries
Historical Perspectives on Apple Production: Fruit Tree Pest Management, Regulation and New Insecticidal Chemistries. Peter Jentsch Extension Associate Department of Entomology Cornell University's Hudson Valley Lab 3357 Rt. 9W; PO box 727 Highland, NY 12528 email: [email protected] Phone 845-691-7151 Mobile: 845-417-7465 http://www.nysaes.cornell.edu/ent/faculty/jentsch/ 2 Historical Perspectives on Fruit Production: Fruit Tree Pest Management, Regulation and New Chemistries. by Peter Jentsch I. Historical Use of Pesticides in Apple Production Overview of Apple Production and Pest Management Prior to 1940 Synthetic Pesticide Development and Use II. Influences Changing the Pest Management Profile in Apple Production Chemical Residues in Early Insect Management Historical Chemical Regulation Recent Regulation Developments Changing Pest Management Food Quality Protection Act of 1996 The Science Behind The Methodology Pesticide Revisions – Requirements For New Registrations III. Resistance of Insect Pests to Insecticides Resistance Pest Management Strategies IV. Reduced Risk Chemistries: New Modes of Action and the Insecticide Treadmill Fermentation Microbial Products Bt’s, Abamectins, Spinosads Juvenile Hormone Analogs Formamidines, Juvenile Hormone Analogs And Mimics Insect Growth Regulators Azadirachtin, Thiadiazine Neonicotinyls Major Reduced Risk Materials: Carboxamides, Carboxylic Acid Esters, Granulosis Viruses, Diphenyloxazolines, Insecticidal Soaps, Benzoyl Urea Growth Regulators, Tetronic Acids, Oxadiazenes , Particle Films, Phenoxypyrazoles, Pyridazinones, Spinosads, Tetrazines , Organotins, Quinolines. 3 I Historical Use of Pesticides in Apple Production Overview of Apple Production and Pest Management Prior to 1940 The apple has a rather ominous origin. Its inception is framed in the biblical text regarding the genesis of mankind. The backdrop appears to be the turbulent setting of what many scholars believe to be present day Iraq. -

US EPA, Pesticide Product Label, LIME-SULFUR SOLUTION
UNITED STATES ENVIRONMENTAL PROTECTION AGENCY WASHINGTON, DC 20460 OFFICE OF CHEMICAL SAFETY AND POLLUTION PREVENTION January 29, 2018 Michael Kellogg Regulatory Consultant c/o Pyxis Regulatory Consulting, Inc. 4110 136th St. Ct NW Gig Harbor, WA 98332 Subject: Notification per PRN 98-10 – New Primary Brand Name. Product Name: Lime-Sulfur Solution Agricultural Fungicide EPA Registration Number: 61842-30 Application Date: 12/21/2017 Decision Number: 537150 Dear Michael Kellogg: The Agency is in receipt of your Application for Pesticide Notification under Pesticide Registration Notice (PRN) 98-10 for the above referenced product. The Registration Division (RD) has conducted a review of this request for its applicability under PRN 98-10 and finds that the action requested falls within the scope of PRN 98-10. The label submitted with the application has been stamped “Notification” and will be placed in our records. Should you wish to add/retain a reference to the company’s website on your label, then please be aware that the website becomes labeling under the Federal Insecticide Fungicide and Rodenticide Act and is subject to review by the Agency. If the website is false or misleading, the product would be misbranded and unlawful to sell or distribute under FIFRA section 12(a)(1)(E). 40 CFR 156.10(a)(5) list examples of statements EPA may consider false or misleading. In addition, regardless of whether a website is referenced on your product’s label, claims made on the website may not substantially differ from those claims approved through the registration process. Therefore, should the Agency find or if it is brought to our attention that a website contains false or misleading statements or claims substantially differing from the EPA approved registration, the website will be referred to the EPA’s Office of Enforcement and Compliance. -

Limestone Resources of Western Washington
State of Washington DANIEL J. EVANS, Governor Department of Conservation H. MA URI CE AHLQUIST, Director DIVISION OF MINES AND GEOLOGY MARSHALL T. HUNTTING, Supervisor Bulletin No. 52 LIMESTONE RESOURCES OF WESTERN WASHINGTON By WILBERT R. DANNER With a section on the UME MOUNTAIN DEPOSIT By GERALD W. THORSEN STATII PRINTING PLANT, OLYMPI A, WASH, 1966 For sale by Department of Conservation, Olympia, Washington. Price, $4,50 FOREWORD Since the early days of Washington's statehood, limestone has been recognized as one of the important mineral resources _of the State. The second annual report of the Washington Geological Survey, published in 1903, gave details on the State's limestone deposits, and in later years five other reports published by the Survey and its successor agencies hove given additional information on this resource. Still other reports by Federal and private agencies hove been published in response to demands for data on limestone here. Although some of the earlier reports included analyses to show the purity of the rocks, very few of the samples for analysis were taken systemati cally in a way that would fairly represent the deposits sampled. Prior to 1900 limestone was produced for use as building stone here, and another important use was for the production of burned Ii me . Portland cement plants soon became leading consumers of Ii mestone, and they con tinue as such to the present time . Limestone is used in large quantities in the pulp industry in the Northwest, and in 1966 there was one commercial lime-burning plant in the State. Recognizing the potential for industrial development in Washington based on more intensive use of our mineral resources, and recognizing the need to up-dote the State's knowledge of raw material resources in order to channel those resources into the State's growing economy, the Industrial Row Materials Advisory Committee of the Deportment of Commerce and Economic Development in 1958 recommended that a comprehensive survey be made of the limestone resources of Washington. -

A Fundamental Study of the Alkaline Sulfide Leaching of Gold
The European Journal of Mineral Processing and Environmental Protection Vol.3, No.3, 1303-0868, 2003, pp. 336-343 A fundamental study of the alkaline sulfide leaching of gold M.I. Jeffrey1* and C.G Anderson2 1 Department of Chemical Engineering, Monash University, Vic., 3800, Australia 2 The Center for Advanced Mineral and Metallurgical Processing, Montana Tech of the University of Montana, Butte, Montana, 59701, USA Received 19 September 2002; accepted 23 September 2003 ABSTRACT Due to the increasing concerns over the emission of sulfur dioxide from roasting and smelting, there has been an increased interest in pressure oxidation as a means of treating gold bearing ores and concentrates. One of the problems with the partial oxidation of the sulfide host matrix to form elemental sulfur is that sulfur containing streams are difficult to treat using cyanidation. In the present paper, the alkaline sulfide system was studied as an alternative to cyanide for recovering gold from elemental sulfur. It was found that the dominant lixiviant for gold in this system is sulfide, and polysulfide was ascertained to be the oxidant. The leaching reaction was determined to be chemically controlled, thus highly dependent on temperature. Other variables investigated include the effect of pH, gold purity, and the ratio of sulfide to polysulfides. It was found that each of these variables had a large impact on the system. © 2003 SDU. All rights reserved. Keywords: Hydrometallurgy; Leaching; Gold 1. INTRODUCTION Many gold ores are problematic as they contain metal sulfides, such as pyrite, arsenopyrite and chalcopyrite. These ores are often refractory, as the gold is encapsulated within the sulfide matrix. -

Nibco Chemical Resistance Guide for Valves & Fittings
NIBCO CHEMICAL RESISTANCE GUIDE FOR VALVES & FITTINGS INTRODUCTION liquids into the system. ABS-DWV is very resistant to a wide vari- ety of materials ranging from sewage to commercial household This chemical resistance guide has been compiled to assist the chemical formulations. ABS-DWV is joined by solvent cementing piping system designer in selecting chemical resistant or threading and can easily be connected to steel, copper, or cast materials. The information given is intended as a guide only. Many iron through the use of transition fittings. conditions can affect the material choices. Careful CPVC — Chlorinated Polyvinyl Chloride Class 23447-B, formerly consideration must be given to temperature, pressure and chemi- designated Type IV, Grade 1 conforming to ASTM D-1784, has cal concentrations before a final material can be selected. physical properties at 73°F similar to those of PVC, and its chemi- Thermoplastics’ and elastomers’ physical characteristics are more cal resistance is similar to or generally better than that of PVC. sensitive to temperature than metals. For this reason, a rating chart CPVC, with a design stress of 2000 psi and maximum service has been developed for each. temperature of 210°F, has proven to be an excellent material for MATERIAL RATINGS hot corrosive liquids, hot or cold water distribution, and similar FOR THERMOPLASTICS & ELASTOMERS applications above the temperature range of PVC. CPVC is joined by solvent cementing, threading or flanging. Temp. in °F = “A” rating, maximum temperature which is recommended, resistant under normal PP (Polypropylene) — Type 1 Polypropylene is a polyolefin, which conditions is lightweight and generally high in chemical resistance. -

SEELEVEL ANNIHILATOR TM Tank Truck Level Gauge
SEELEVEL ANNIHILATOR TM Tank Truck Level Gauge MODEL 806-B / 806-Bi MANUAL Includes version 2.03 software information CANADA USA garnetinstruments.com Garnet Instruments Garnet US Inc. 286 Kaska Road 5360 Old Granbury Road 1-800-617-7384 Sherwood Park, AB T8A 4G7 Granbury, TX 76049 GARNET SEELEVEL ANNIHILATOR TM Tank Truck Level Gauge MODEL 806-B / 806-Bi Includes version 2.03 software information Table of Contents CHAPTER 1 - OVERVIEW .............................................................................................3 CHAPTER 2 - NEW FEATURES OF THE ANNIHILATOR .....................................4 CHAPTER 3 - GAUGE DESCRIPTION .......................................................................5 CHAPTER 4 - UNIQUE FEATURES ............................................................................9 CHAPTER 5 - SENDER BAR LIMITS OF RESISTIVITY ........................................ 10 CHAPTER 6 - INSTALLATION GUIDE ..................................................................... 13 CHAPTER 7 - DISPLAY PROGRAMMING INSTRUCTIONS ............................ 20 CHAPTER 8 - TROUBLESHOOTING GUIDE ......................................................... 27 CHAPTER 9 - SPECIFICATIONS ............................................................................... 29 CHAPTER 10 - SERVICE AND WARRANTY INFORMATION.......................... 30 806-B/806-Bi Manual_v2.9 - 11-Dec-2019 Page 2 806-B/806-Bi Manual CHAPTER 1 - OVERVIEW ongratulations on purchasing the Garnet Instruments Model C806-B or 806-Bi SEELEVEL ANNIHILATOR -

Chemical Studies of the Lime-Sulfur Lead Arsenate Spray Mixture
Research Bulletin No. 12 June, 1913 Chemical Studies of the Lime-Sulfur Lead Arsenate Spray Mixture BY W. E. RUTH AGRICULTURAL EXPERIMENT STATION IOWA STATE COLLEGE OF AGRICULTURE AND THE MECHANIC ARTS CHEMICAL SECTION A MES, IOWA SUMMARY l\lixing lead arsenate with lime sulfur results Ill: 1. A decrease of sulfur in solution. 2. A decrease of calcium in solution. 3. Increase of thiosulfate in solution. 4. Increase of thiosulfate in the residue. 5. Increase of sulfite in solution. 6. Formation of lead sulfide. 7. Formation of a compound containing arsenic and sulfur but insolnable in the lime-sulfur solution, probably lead thioarsenate. CHEMICAL STUDIES OF THE LIME SULFUR--LEAD ARSENATE SPRAY MIXTURE BY W. E. RUTH INTRODUCTORY. The use of lime-sulfur as an orchard spray originated with 1\1. F. Duseyl of Fresno, California. It had been previously used as a sheep dip for killing ticks, but Mr. Dusey tried it successfully as a means of combating San Jose scale. Some years later San Jose scale was discovered in different fruit districts in the middle Atlantic states and after a time the lime-sulfur treatment came to be recognized there, as well as in California, as the best means for combating that insect. At that time bordeaux mixture was regarded as the most efficient spray for control of fungus a;seases in orchard8 rf deciduous fruits, and to control leaf ea1 in~ iments also. it was the common practice to combine with bordeaux mixture some arsenical poison. It was early recognized that lime-sulfur pos sessed some fungicidal properties although it had not yet made a place for itself as a standard fungicide. -

Recommendation Guidebook
SOIL TEST RECOMMENDATIONS for VIRGINIA Prepared by: R. O. Maguire, Professor and Extension Specialist S. E. Heckendorn, Soil Testing Laboratory Manager September 2021 ii TABLE OF CONTENTS Page I. INTRODUCTION ....................................................................................................... 1 II. COMPUTER CROP CODE NUMBERS ................................................................... 2 III. EXTENSION UNIT CODE LIST ............................................................................... 5 IV. SOIL TEST METHODS and CALIBRATIONS ........................................................ 6 A. Tests Offered and Methods Used ......................................................................... 6 B. Soil Test Calibrations ........................................................................................... 7 C. Critical Soil Test Levels ....................................................................................... 11 V. FERTILIZER RECOMMENDATIONS - GENERAL ............................................... 12 A. How Recommendations Are Made ...................................................................... 12 B. N Allowance for Use of Legumes ........................................................................ 12 VI. FERTILIZER RECOMMENDATIONS - N, P, K - BY CROP ................................. 15 A. Commercial Crop Production .............................................................................. 16 1. Field Crops .............................................................................................. -

Benkaraache, Sanae.Pdf
Université du Québec Institut National de la Recherche Scientifique Eau, Terre et Environnement ÉVALUATION DES POSSIBILITÉS DU TRAITEMENT D’UN SOL CONTAMINÉ PAR DU CUIVRE ET DU NICKEL PROVENANT DE RÉSIDUS MINIERS EN UTILISANT DES PROCÉDÉS PHYSIQUE ET CHIMIQUE Préparé par Sanae BENKARAACHE Mémoire présenté Pour l’obtention du grade de Maître ès en Sciences de l’Eau Jury d’évaluation Examinateur externe Monsieur Mostafa EL FAHIME ISHÉDD, Maroc Examinateur externe Monsieur Antoine KARAM Université Laval Examinateur interne Monsieur Rajeshwar Dayal TYAGI INRS-ETE, Université du Québec Directeur de recherche Monsieur Guy MERCIER INRS-ETE, Université du Québec Codirecteur de recherche Monsieur Jean-François BLAIS INRS-ETE, Université du Québec © Droits réservés à Sanae BENKARAACHE, 2016 RÉSUMÉ Le but de ce projet est d’explorer les possibilités de réhabilitation d’un sol contaminé à la fois par des hydrocarbures pétroliers et des métaux lourds. Néanmoins, l’étude s’est plus focalisée sur les contaminants métalliques provenant de résidus miniers chargés en cuivre et en nickel. Le sol contaminé est classé selon les cinq fractions suivantes : f1 > 4 mm ; f2 1-2 mm ; f3 0,250-1 mm et f4 <0,250 mm et a subi une caractérisation par dissolution acide puis ICP-AES, diffraction des rayons X (DRX) et microscope électronique à balayage (MEB). D’après les analyses, les teneurs respectives sont de l’ordre de 2102 mg Cu/kg et 1146 mg Ni/kg pour f4, ce qui dépasse le critère C du Ministère du Développement Durable de l’Environnement et de la Lutte contre les Changements Climatiques (MDDELCC). -

(12) Patent Application Publication (10) Pub. No.: US 2011/0262343 A1 Hojjatie Et Al
US 2011 O2623.43A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2011/0262343 A1 Hojjatie et al. (43) Pub. Date: Oct. 27, 2011 (54) PROCESS FOR PREPARATION OF CALCIUM Publication Classification THIOSULFATE LIQUID SOLUTION FROM (51) Int. Cl. LIME, SULFUR, AND SULFUR DIOXIDE COB I7/64 (2006.01) (52) U.S. Cl. ........................................................ 423/514 (75) Inventors: Michael Massoud Hojjatie, Tucson, AZ (US); Yelena Feinstein, (57) ABSTRACT Tucson, AZ (US); Constance Lynn An efficient batch or semi-continuous method of production Frank Lockhart, Tucson, AZ (US) of relative high concentration, low byproduct-containing, cal cium thiosulfate (CaSO) from lime, sulfur or calcium (73) Assignee: TESSENDERLO KERLEY, INC., polysulfide, and sulfur dioxide under elevated reaction tem perature conditions is described. The product is an emulsion Phoenix, AZ (US) of liquid calcium thiosulfate and solid byproducts. Under the conditions of the art, including the mole ratios of lime to (21) Appl. No.: 12/764,843 sulfur, the temperature of the reaction process and the sulfur dioxide reaction conditions, including rate and duration, the (22) Filed: Apr. 21, 2010 byproducts are reduced to about 2% by weight. Recycled Sulfur Calcium Thiosulfate ---O-G)-GED-G-G) Patent Application Publication Oct. 27, 2011 Sheet 1 of 4 US 2011/0262343 A1 PROCESS KINETICS FORCATS PREPARATION FROM "TRUE CALCUM POLYSULFIDE" AND SO2 PHASA FUNCTION OF TIME l2 pro- p- --------------------- ... ------ --- -- - - -- m-- --- 3. 8 . se: ... "aggers. -x x w w w x * v- w r - - - - - - - - - - - im w w w w w w w w - Y, 1 it 6 --------------------------------------- s w \r --------- ---------- ry. & : 4 - - - - -- - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - sex Xx .. -

Chemical Resistance Guide
CATALOG C-CRG-0517 VALVES Pressure-rated bronze, iron and alloy-iron gate, globe and check valves • Pressure- rated bronze ball valves • Boiler specialty valves • Commercial and industrial butterfly valves • Lined butterfly valves • Circuit balancing valves and kits • Carbon and stainless steel ball valves • ANSI flanged steel ball valves • Lined ball valves • Pneumatic and electric actuators and controls • Grooved ball and butterfly valves • High performance butterfly valves • UL/FM fire protection valves • MSS specification valves • Bronze specialty valves • Low pressure gate, globe, -GU check and ball valves • Frostproof sillcocks • Quarter-turn supply stops • Quarter- EM ID turn low pressure valves • PVC and CPVC plumbing and industrial ball valves • H Bronze and iron y-strainers • Sample valves • Sanitary valves • Lead-free valves E • Hydronic valves • Labor saving valves • Manifold systems • Water temperature C control valves • System quality valves • Press x PEX transition valves FITTINGS Wrot and cast copper pressure and drainage fittings • Cast copper alloy flanges • Powder coated steel companion flanges • Wrot and cast press fittings • ABS and PVC DWV fittings • Schedule 40 PVC pressure fittings • CPVC CTS fittings • CPVC CTS-to-metal transition fittings • Schedule 80 PVC and CPVC systems • Lead-free fittings • Press x PEX transition fittings • Cast bronze push fittings LEAD-FREE: Weighted average lead content ≤0.25% FLEXIBLE PIPING SYSTEMS PE-RT and PEX tubing for potable and radiant applications • Insulated tubing • Risers