Kcnj5 Mutations and Adrenocortical Cell Growth
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Mineralocorticoid Receptor Leads to Increased Expression of EGFR
www.nature.com/scientificreports OPEN The mineralocorticoid receptor leads to increased expression of EGFR and T‑type calcium channels that support HL‑1 cell hypertrophy Katharina Stroedecke1,2, Sandra Meinel1,2, Fritz Markwardt1, Udo Kloeckner1, Nicole Straetz1, Katja Quarch1, Barbara Schreier1, Michael Kopf1, Michael Gekle1 & Claudia Grossmann1* The EGF receptor (EGFR) has been extensively studied in tumor biology and recently a role in cardiovascular pathophysiology was suggested. The mineralocorticoid receptor (MR) is an important efector of the renin–angiotensin–aldosterone‑system and elicits pathophysiological efects in the cardiovascular system; however, the underlying molecular mechanisms are unclear. Our aim was to investigate the importance of EGFR for MR‑mediated cardiovascular pathophysiology because MR is known to induce EGFR expression. We identifed a SNP within the EGFR promoter that modulates MR‑induced EGFR expression. In RNA‑sequencing and qPCR experiments in heart tissue of EGFR KO and WT mice, changes in EGFR abundance led to diferential expression of cardiac ion channels, especially of the T‑type calcium channel CACNA1H. Accordingly, CACNA1H expression was increased in WT mice after in vivo MR activation by aldosterone but not in respective EGFR KO mice. Aldosterone‑ and EGF‑responsiveness of CACNA1H expression was confrmed in HL‑1 cells by Western blot and by measuring peak current density of T‑type calcium channels. Aldosterone‑induced CACNA1H protein expression could be abrogated by the EGFR inhibitor AG1478. Furthermore, inhibition of T‑type calcium channels with mibefradil or ML218 reduced diameter, volume and BNP levels in HL‑1 cells. In conclusion the MR regulates EGFR and CACNA1H expression, which has an efect on HL‑1 cell diameter, and the extent of this regulation seems to depend on the SNP‑216 (G/T) genotype. -

Next Generation Sequencing for Molecular Confirmation of Hereditary
Arch Cardiol Mex. 2015;85(1):68---72 www.elsevier.com.mx SPECIAL ARTICLE Next generation sequencing for molecular confirmation of hereditary sudden cardiac death syndromes a,∗ b b b Manlio F. Márquez , David Cruz-Robles , Selene Ines-Real , Gilberto Vargas-Alarcón , a Manuel Cárdenas a Departamento de Electrofisiología, Instituto Nacional de Cardiología Ignacio Chávez, México, D.F., Mexico b Departamento de Biología Molecular, Instituto Nacional de Cardiología Ignacio Chávez, México, D.F., Mexico Received 26 March 2014; accepted 8 December 2014 KEYWORDS Abstract Hereditary sudden cardiac death syndromes comprise a wide range of diseases result- Arrhythmias; ing from alteration in cardiac ion channels. Genes involved in these syndromes represent diverse Hereditary sudden mutations that cause the altered encoding of the diverse proteins constituting these channels, cardiac death thus affecting directly the currents of the corresponding ions. In the present article we will syndromes; briefly review how to arrive to a clinical diagnosis and we will present the results of molecular Right ventricle genetic studies made in Mexican subjects attending the SCD Syndromes Clinic of the National arrhythmogenic Institute of Cardiology of Mexico City. cardiomyopathy; © 2014 Instituto Nacional de Cardiología Ignacio Chávez. Published by Masson Doyma México Brugada syndrome S.A. All rights reserved. PALABRAS CLAVE Confirmación diagnóstica molecular mediante secuenciación masiva de nueva Arritmias; generación (‘‘next generation sequencing’’) en síndromes hereditarios de muerte Síndromes súbita cardíaca hereditarios de Resumen Los síndromes hereditarios de muerte súbita cardíaca comprenden una amplia gama muerte súbita; Displasia de enfermedades resultantes de la alteración en los canales iónicos cardíacos. Los genes implicados en estos síndromes presentan mutaciones que causan alteraciones de las diversas Arritmogénica del proteínas que constituyen estos canales y que, por lo tanto, afectan directamente a las difer- ventriculo derecho; entes corrientes iónicas. -

Investigating Unexplained Deaths for Molecular Autopsies
The author(s) shown below used Federal funding provided by the U.S. Department of Justice to prepare the following resource: Document Title: Investigating Unexplained Deaths for Molecular Autopsies Author(s): Yingying Tang, M.D., Ph.D, DABMG Document Number: 255135 Date Received: August 2020 Award Number: 2011-DN-BX-K535 This resource has not been published by the U.S. Department of Justice. This resource is being made publically available through the Office of Justice Programs’ National Criminal Justice Reference Service. Opinions or points of view expressed are those of the author(s) and do not necessarily reflect the official position or policies of the U.S. Department of Justice. Final Technical Report NIJ FY 11 Basic Science Research to Support Forensic Science 2011-DN-BX-K535 Investigating Unexplained Deaths through Molecular Autopsies May 28, 2017 Yingying Tang, MD, PhD, DABMG Principal Investigator Director, Molecular Genetics Laboratory Office of Chief Medical Examiner 421 East 26th Street New York, NY, 10016 Tel: 212-323-1340 Fax: 212-323-1540 Email: [email protected] Page 1 of 41 This resource was prepared by the author(s) using Federal funds provided by the U.S. Department of Justice. Opinions or points of view expressed are those of the author(s) and do not necessarily reflect the official position or policies of the U.S. Department of Justice. Abstract Sudden Unexplained Death (SUD) is natural death in a previously healthy individual whose cause remains undetermined after scene investigation, complete autopsy, and medical record review. SUD affects children and adults, devastating families, challenging medical examiners, and is a focus of research for cardiologists, neurologists, clinical geneticists, and scientists. -

Cardiogenetics Testing Reference Guide December 2018
Cardiogenetics Testing reference guide December 2018 Why Choose Ambry More than 1 in 200 people have an inherited cardiovascular condition. Ambry’s mission is to provide the most advanced genetic testing information available to help you identity those at-risk and determine the best treatment options. If we know a patient has a disease-causing genetic change, not only does it mean better disease management, it also indicates that we can test others in the family and provide them with potentially life-saving information. Diseases and Testing Options cardiomyopathies arrhythmias Hypertrophic Cardiomyopathy (HCMNext) Catecholaminergic Polymorphic Ventricular Dilated Cardiomyopathy (DCMNext) Tachycardia (CPVTNext) Arrhythmogenic Right Ventricular Long QT Syndrome, Short QT Syndrome, Cardiomyopathy (ARVCNext) Brugada Syndrome (LongQTNext, RhythmNext) Cardiomyopathies (CMNext, CardioNext) Arrhythmias (RhythmNext, CardioNext) other cardio conditions Transthyretin Amyloidosis (TTR) familial hypercholesterolemia Noonan Syndrome (NoonanNext) and lipid disorders Hereditary Hemorrhagic Telangiectasia Familial Hypercholesterolemia (FHNext) (HHTNext) Sitosterolemia (Sitosterolemia Panel) Comprehensive Lipid Menu thoracic aortic aneurysms (CustomNext-Cardio) and dissections Familial Chylomicronemia Syndrome (FCSNext) Thoracic Aneurysms and Dissections, aortopathies (TAADNext) Marfan Syndrome (TAADNext) Ehlers-Danlos Syndrome (TAADNext) Targeted Panels Gene Comparison ALL PANELS HAVE A TURNAROUND TIME OF 2-3 WEEKS arrhythmias CPVTNext CPVTNext CASQ2, -

Primary Aldosteronism Diagnostics: KCNJ5 Mutations and Hybrid Steroid Synthesis in Aldosterone-Producing Adenomas
13 Review Article Primary aldosteronism diagnostics: KCNJ5 mutations and hybrid steroid synthesis in aldosterone-producing adenomas Juilee Rege1, Adina F. Turcu2, William E. Rainey1,2 1Department of Molecular and Integrative Physiology, 2Division of Metabolism, Endocrinology, and Diabetes, Department of Internal Medicine, University of Michigan, Ann Arbor, MI, USA Contributions: (I) Conception and design: J Rege, WE Rainey; (II) Administrative support: WE Rainey; (III) Provision of study materials or patients: None; (IV) Collection and assembly of data: J Rege, WE Rainey; (V) Data analysis and interpretation: None; (VI) Manuscript writing: All authors; (VII) Final approval of manuscript: All authors. Correspondence to: William E. Rainey, PhD. Departments of Molecular and Integrative Physiology and Internal Medicine, University of Michigan, Ann Arbor, MI 48109, USA. Email: [email protected]. Abstract: Primary aldosteronism (PA) is characterized by autonomous aldosterone production by renin- independent mechanisms and is most commonly sporadic. While 60–70% of sporadic PA can be attributed to bilateral hyperaldosteronism, the remaining 30–40% is caused by a unilateral aldosterone-producing adenoma (APA). Somatic mutations in or near the selectivity filter the KCNJ5 gene (encoding the potassium channel GIRK4) have been implicated in the pathogenesis of both sporadic and familial PA. Several studies using tumor tissue, peripheral and adrenal vein samples from PA patients have demonstrated that along with aldosterone, the hybrid steroids 18-hydroxycortisol (18OHF) and 18-oxocortisol (18oxoF) are a hallmark of APA harboring KCNJ5 mutations. Herein, we review the recent advances with respect to the molecular mechanisms underlying the pathogenesis of PA and the steroidogenic fingerprints of KCNJ5 mutations. In addition, we present an outlook toward the future of PA subtyping and diagnostic work-up utilizing steroid profiling. -

Spatial Distribution of Leading Pacemaker Sites in the Normal, Intact Rat Sinoa
Supplementary Material Supplementary Figure 1: Spatial distribution of leading pacemaker sites in the normal, intact rat sinoatrial 5 nodes (SAN) plotted along a normalized y-axis between the superior vena cava (SVC) and inferior vena 6 cava (IVC) and a scaled x-axis in millimeters (n = 8). Colors correspond to treatment condition (black: 7 baseline, blue: 100 µM Acetylcholine (ACh), red: 500 nM Isoproterenol (ISO)). 1 Supplementary Figure 2: Spatial distribution of leading pacemaker sites before and after surgical 3 separation of the rat SAN (n = 5). Top: Intact SAN preparations with leading pacemaker sites plotted during 4 baseline conditions. Bottom: Surgically cut SAN preparations with leading pacemaker sites plotted during 5 baseline conditions (black) and exposure to pharmacological stimulation (blue: 100 µM ACh, red: 500 nM 6 ISO). 2 a &DUGLDFIoQChDQQHOV .FQM FOXVWHU &DFQDG &DFQDK *MD &DFQJ .FQLS .FQG .FQK .FQM &DFQDF &DFQE .FQM í $WSD .FQD .FQM í .FQN &DVT 5\U .FQM &DFQJ &DFQDG ,WSU 6FQD &DFQDG .FQQ &DFQDJ &DFQDG .FQD .FQT 6FQD 3OQ 6FQD +FQ *MD ,WSU 6FQE +FQ *MG .FQN .FQQ .FQN .FQD .FQE .FQQ +FQ &DFQDD &DFQE &DOP .FQM .FQD .FQN .FQG .FQN &DOP 6FQD .FQD 6FQE 6FQD 6FQD ,WSU +FQ 6FQD 5\U 6FQD 6FQE 6FQD .FQQ .FQH 6FQD &DFQE 6FQE .FQM FOXVWHU V6$1 L6$1 5$ /$ 3 b &DUGLDFReFHSWRUV $GUDF FOXVWHU $GUDD &DY &KUQE &KUP &KJD 0\O 3GHG &KUQD $GUE $GUDG &KUQE 5JV í 9LS $GUDE 7SP í 5JV 7QQF 3GHE 0\K $GUE *QDL $QN $GUDD $QN $QN &KUP $GUDE $NDS $WSE 5DPS &KUP 0\O &KUQD 6UF &KUQH $GUE &KUQD FOXVWHU V6$1 L6$1 5$ /$ 4 c 1HXURQDOPURWHLQV -
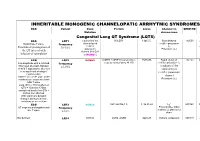
Inheritable Arrhytmia Syndromes
INHERITABLE MONOGENIC CHANNELOPATIC ARRHYTMIC SYNDROMES ECG Variant Gene Protein Locus Channel on OMIM NO IP Mutation chromosome Congenital Long QT Syndrome (LQTS) ECG LQT1 α subunit of the KvLQT1 11p15.5. Slow delayed 192500 AD slow delayed rectifier potassium or Broad base T wave. Frequency rectifier I AR Paradoxical prolongation of 30-35% ks potassium Potassium (IKs) the QT interval with channel (KvLQT1 infusion of epinephrine or KCNQ1) ECG LQT2 KCNH2 (HERG + MiRP1) human ether- 7q35q36 Rapid delayed 152.427 AD a-go-go related gene HERG rectifier potassium I Low-amplitude with a notched, Frequency kr bifurcated alternant, biphasic 25-30% α subunit of the or bifid T appearance due to a rapid delayed very significant slowing of rectifier potassium repolarization. channel KCNH2 on L413P and L559H mutations are associated with Potassium (IKr) bifid T wave Long. QTc > 470ms affected QTc = 450ms to 470ms considered border line QTc < 450ms non affected. With significant dynamic changes during heart rate variations or on exertion + ECG LQT3 SCN5A hH1 and NaV1.5 3, 3p 21-24 INa 600163 AD Prolonged I + influx ST segment prolongation and Frequency Na late T wave. in phase 2, plateau or 5-10% dome Not defined LQT4 KCNJ2 ANK2, ANKB 4q25-27 Sodium, potassium 600919 AR and calcium + Not defined LQT5 KCNE1 minK K Efflux 176261 AD Not defined LQT6 KCNE2 MiRP1 603796 MiRP1 Frequency rare Modest prolongation of QT LQT7 Andersen KCNJ2 Kir2.1 Ik1 Reduction 170390 AD interval, prominent U wave, Great reduction in I Tawil-syndrome k1 frequent PVCs, PVT, result in the bidirectional VT. -

Identification of Key Pathways and Genes in Dementia Via Integrated Bioinformatics Analysis
bioRxiv preprint doi: https://doi.org/10.1101/2021.04.18.440371; this version posted July 19, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Identification of Key Pathways and Genes in Dementia via Integrated Bioinformatics Analysis Basavaraj Vastrad1, Chanabasayya Vastrad*2 1. Department of Biochemistry, Basaveshwar College of Pharmacy, Gadag, Karnataka 582103, India. 2. Biostatistics and Bioinformatics, Chanabasava Nilaya, Bharthinagar, Dharwad 580001, Karnataka, India. * Chanabasayya Vastrad [email protected] Ph: +919480073398 Chanabasava Nilaya, Bharthinagar, Dharwad 580001 , Karanataka, India bioRxiv preprint doi: https://doi.org/10.1101/2021.04.18.440371; this version posted July 19, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract To provide a better understanding of dementia at the molecular level, this study aimed to identify the genes and key pathways associated with dementia by using integrated bioinformatics analysis. Based on the expression profiling by high throughput sequencing dataset GSE153960 derived from the Gene Expression Omnibus (GEO), the differentially expressed genes (DEGs) between patients with dementia and healthy controls were identified. With DEGs, we performed a series of functional enrichment analyses. Then, a protein–protein interaction (PPI) network, modules, miRNA-hub gene regulatory network and TF-hub gene regulatory network was constructed, analyzed and visualized, with which the hub genes miRNAs and TFs nodes were screened out. Finally, validation of hub genes was performed by using receiver operating characteristic curve (ROC) analysis. -

Cardiovascular Diseases Genetic Testing Program Information
Cardiovascular Diseases Genetic Testing Program Description: Congenital Heart Disease Panels We offer comprehensive gene panels designed to • Congenital Heart Disease Panel (187 genes) diagnose the most common genetic causes of hereditary • Heterotaxy Panel (114 genes) cardiovascular diseases. Testing is available for congenital • RASopathy/Noonan Spectrum Disorders Panel heart malformation, cardiomyopathy, arrythmia, thoracic (31 genes) aortic aneurysm, pulmonary arterial hypertension, Marfan Other Panels syndrome, and RASopathy/Noonan spectrum disorders. • Pulmonary Arterial Hypertension (PAH) Panel Hereditary cardiovascular disease is caused by variants in (20 genes) many different genes, and may be inherited in an autosomal dominant, autosomal recessive, or X-linked manner. Other Indications: than condition-specific panels, we also offer single gene Panels: sequencing for any gene on the panels, targeted variant • Confirmation of genetic diagnosis in a patient with analysis, and targeted deletion/duplication analysis. a clinical diagnosis of cardiovascular disease Tests Offered: • Carrier or pre-symptomatic diagnosis identification Arrythmia Panels in individuals with a family history of cardiovascular • Comprehensive Arrhythmia Panel (81 genes) disease of unknown genetic basis • Atrial Fibrillation (A Fib) Panel (28 genes) Gene Specific Sequencing: • Atrioventricular Block (AV Block) Panel (7 genes) • Confirmation of genetic diagnosis in a patient with • Brugada Syndrome Panel (21 genes) cardiovascular disease and in whom a specific -

Mutations of Voltage-Gated Ionic Channels and Risk of Severe Cardiac Arrhythmias
Acta Cardiol Sin 2019;35:99-110 Review Article doi: 10.6515/ACS.201903_35(2).20181028A Mutations of Voltage-Gated Ionic Channels and Risk of Severe Cardiac Arrhythmias Amir Dehghani-Samani,1 Samin Madreseh-Ghahfarokhi2 and Azam Dehghani-Samani3 Background: Arrhythmias as important malfunctions of heart are known as abnormal rhythm of heart. Several causes can make arrhythmias and most of them are related to generation and/or conduction of action potential in heart. Action potential in myocytes results from the sequential opening and closing of ion channel proteins that span the plasma membrane of individual myocytes. Action potential’s conduction through the heart is depended on electrical coupling between myocytes, which is mediated by gap junctions. Generation and conduction of action potentials are related to perfect action of ionic channels in heart. Objectives: This novel review comprehensively addressed the ionic mechanisms of the arrhythmogenic mutations in cardiac voltage-gated ionic channels including: CACNA1C, CACNA1D, KCNA5, KCND2, KCND3, KCNE1, KCNE2, KCNE5, KCNH2, KCNJ2, KCNJ5, KCNQ1, SCN4A, SCN5A, SCN1B, SCN2B, SCN3B and SCN4B. Methods: Current study, for the first time, review and discuses about relation between cardiac arrhythmias and whole of important voltage gated ionic channels from different families, altogether and at the same time. Results: This review clears that mutations in voltage-gated ionic channels play important roles in generation of severe cardiac arrhythmias, and among them it is looked that mutations in voltage-gated potassium channels are more important. Conclusions: Most of induced arrhythmias due to voltage-gated ionic channels mutations result in action potentials prolongation and long QT syndromes. -

(KCNK3) Channels in the Lung: from Cell Biology to Clinical Implications
REVIEW PULMONARY CIRCULATION AND PHYSIOPATHOLOGY TASK-1 (KCNK3) channels in the lung: from cell biology to clinical implications Andrea Olschewski1,2, Emma L. Veale3, Bence M. Nagy2, Chandran Nagaraj1,2, Grazyna Kwapiszewska1,2, Fabrice Antigny4,5,6, Mélanie Lambert4,5,6, Marc Humbert 4,5,6, Gábor Czirják7, Péter Enyedi7 and Alistair Mathie3 Affiliations: 1Ludwig Boltzmann Institute for Lung Vascular Research Graz, Graz, Austria. 2Institute of Physiology, Medical University of Graz, Graz, Austria. 3Medway School of Pharmacy, University of Kent, Central Avenue, Chatham Maritime, UK. 4Univ. Paris-Sud, Faculté de Médecine, Kremlin-Bicêtre, France. 5AP-HP, Centre de Référence de l’Hypertension Pulmonaire Sévère, Département Hospitalo-Universitaire (DHU) Thorax Innovation, Service de Pneumologie et Réanimation Respiratoire, Hôpital de Bicêtre, Le Kremlin- Bicêtre, France. 6UMRS 999, INSERM and Univ. Paris–Sud, Laboratoire d’Excellence (LabEx) en Recherche sur le Médicament et l’Innovation Thérapeutique (LERMIT), Hôpital-Marie-Lannelongue, Le Plessis Robinson, France. 7Dept of Physiology, Semmelweis University, Budapest, Hungary. Correspondence: Andrea Olschewski, Ludwig Boltzmann Institute for Lung Vascular Research, Stiftingtalstrasse 24, Graz-8010, Austria. E-mail: [email protected] @ERSpublications Current advancements of TASK-1/KCNK3 channels in the human pulmonary circulation in health and disease http://ow.ly/xgJo30fNZRN Cite this article as: Olschewski A, Veale EL, Nagy BM, et al. TASK-1 (KCNK3) channels in the lung: from cell biology to clinical implications. Eur Respir J 2017; 50: 1700754 [https://doi.org/10.1183/ 13993003.00754-2017]. ABSTRACT TWIK-related acid-sensitive potassium channel 1 (TASK-1 encoded by KCNK3) belongs to the family of two-pore domain potassium channels. -

Potassium Channels and Their Potential Roles in Substance Use Disorders
International Journal of Molecular Sciences Review Potassium Channels and Their Potential Roles in Substance Use Disorders Michael T. McCoy † , Subramaniam Jayanthi † and Jean Lud Cadet * Molecular Neuropsychiatry Research Branch, NIDA Intramural Research Program, Baltimore, MD 21224, USA; [email protected] (M.T.M.); [email protected] (S.J.) * Correspondence: [email protected]; Tel.: +1-443-740-2656 † Equal contributions (joint first authors). Abstract: Substance use disorders (SUDs) are ubiquitous throughout the world. However, much re- mains to be done to develop pharmacotherapies that are very efficacious because the focus has been mostly on using dopaminergic agents or opioid agonists. Herein we discuss the potential of using potassium channel activators in SUD treatment because evidence has accumulated to support a role of these channels in the effects of rewarding drugs. Potassium channels regulate neuronal action potential via effects on threshold, burst firing, and firing frequency. They are located in brain regions identified as important for the behavioral responses to rewarding drugs. In addition, their ex- pression profiles are influenced by administration of rewarding substances. Genetic studies have also implicated variants in genes that encode potassium channels. Importantly, administration of potassium agonists have been shown to reduce alcohol intake and to augment the behavioral effects of opioid drugs. Potassium channel expression is also increased in animals with reduced intake of methamphetamine. Together, these results support the idea of further investing in studies that focus on elucidating the role of potassium channels as targets for therapeutic interventions against SUDs. Keywords: alcohol; cocaine; methamphetamine; opioids; pharmacotherapy Citation: McCoy, M.T.; Jayanthi, S.; Cadet, J.L.