(ASD) and Intellectual Disability
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Analysis of the Indacaterol-Regulated Transcriptome in Human Airway
Supplemental material to this article can be found at: http://jpet.aspetjournals.org/content/suppl/2018/04/13/jpet.118.249292.DC1 1521-0103/366/1/220–236$35.00 https://doi.org/10.1124/jpet.118.249292 THE JOURNAL OF PHARMACOLOGY AND EXPERIMENTAL THERAPEUTICS J Pharmacol Exp Ther 366:220–236, July 2018 Copyright ª 2018 by The American Society for Pharmacology and Experimental Therapeutics Analysis of the Indacaterol-Regulated Transcriptome in Human Airway Epithelial Cells Implicates Gene Expression Changes in the s Adverse and Therapeutic Effects of b2-Adrenoceptor Agonists Dong Yan, Omar Hamed, Taruna Joshi,1 Mahmoud M. Mostafa, Kyla C. Jamieson, Radhika Joshi, Robert Newton, and Mark A. Giembycz Departments of Physiology and Pharmacology (D.Y., O.H., T.J., K.C.J., R.J., M.A.G.) and Cell Biology and Anatomy (M.M.M., R.N.), Snyder Institute for Chronic Diseases, Cumming School of Medicine, University of Calgary, Calgary, Alberta, Canada Received March 22, 2018; accepted April 11, 2018 Downloaded from ABSTRACT The contribution of gene expression changes to the adverse and activity, and positive regulation of neutrophil chemotaxis. The therapeutic effects of b2-adrenoceptor agonists in asthma was general enriched GO term extracellular space was also associ- investigated using human airway epithelial cells as a therapeu- ated with indacaterol-induced genes, and many of those, in- tically relevant target. Operational model-fitting established that cluding CRISPLD2, DMBT1, GAS1, and SOCS3, have putative jpet.aspetjournals.org the long-acting b2-adrenoceptor agonists (LABA) indacaterol, anti-inflammatory, antibacterial, and/or antiviral activity. Numer- salmeterol, formoterol, and picumeterol were full agonists on ous indacaterol-regulated genes were also induced or repressed BEAS-2B cells transfected with a cAMP-response element in BEAS-2B cells and human primary bronchial epithelial cells by reporter but differed in efficacy (indacaterol $ formoterol . -

MAP17's Up-Regulation, a Crosspoint in Cancer and Inflammatory
García-Heredia and Carnero Molecular Cancer (2018) 17:80 https://doi.org/10.1186/s12943-018-0828-7 REVIEW Open Access Dr. Jekyll and Mr. Hyde: MAP17’s up- regulation, a crosspoint in cancer and inflammatory diseases José M. García-Heredia1,2,3 and Amancio Carnero1,3* Abstract Inflammation is a common defensive response that is activated after different harmful stimuli. This chronic, or pathological, inflammation is also one of the causes of neoplastic transformation and cancer development. MAP17 is a small protein localized to membranes with a restricted pattern of expression in adult tissues. However, its expression is common in destabilized cells, as it is overexpressed both in inflammatory diseases and in cancer. MAP17 is overexpressed in most, if not all, carcinomas and in many tumors of mesenchymal origin, and correlates with higher grade and poorly differentiated tumors. This overexpression drives deep changes in cell homeostasis including increased oxidative stress, deregulation of signaling pathways and increased growth rates. Importantly, MAP17 is associated in tumors with inflammatory cells infiltration, not only in cancer but in various inflammatory diseases such as Barret’s esophagus, lupus, Crohn’s, psoriasis and COPD. Furthermore, MAP17 also modifies the expression of genes connected to inflammation, showing a clear induction of the inflammatory profile. Since MAP17 appears highly correlated with the infiltration of inflammatory cells in cancer, is MAP17 overexpression an important cellular event connecting tumorigenesis and inflammation? Keywords: MAP17, Cancer, Inflammatory diseases Background process ends when the activated cells undergo apoptosis in Inflammatory response is a common defensive process acti- a highly regulated process that finishes after pathogens and vated after different harmful stimuli, constituting a highly cell debris have been phagocytized [9]. -

ADHD) Gene Networks in Children of Both African American and European American Ancestry
G C A T T A C G G C A T genes Article Rare Recurrent Variants in Noncoding Regions Impact Attention-Deficit Hyperactivity Disorder (ADHD) Gene Networks in Children of both African American and European American Ancestry Yichuan Liu 1 , Xiao Chang 1, Hui-Qi Qu 1 , Lifeng Tian 1 , Joseph Glessner 1, Jingchun Qu 1, Dong Li 1, Haijun Qiu 1, Patrick Sleiman 1,2 and Hakon Hakonarson 1,2,3,* 1 Center for Applied Genomics, Children’s Hospital of Philadelphia, Philadelphia, PA 19104, USA; [email protected] (Y.L.); [email protected] (X.C.); [email protected] (H.-Q.Q.); [email protected] (L.T.); [email protected] (J.G.); [email protected] (J.Q.); [email protected] (D.L.); [email protected] (H.Q.); [email protected] (P.S.) 2 Division of Human Genetics, Department of Pediatrics, The Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA 3 Department of Human Genetics, Children’s Hospital of Philadelphia, Philadelphia, PA 19104, USA * Correspondence: [email protected]; Tel.: +1-267-426-0088 Abstract: Attention-deficit hyperactivity disorder (ADHD) is a neurodevelopmental disorder with poorly understood molecular mechanisms that results in significant impairment in children. In this study, we sought to assess the role of rare recurrent variants in non-European populations and outside of coding regions. We generated whole genome sequence (WGS) data on 875 individuals, Citation: Liu, Y.; Chang, X.; Qu, including 205 ADHD cases and 670 non-ADHD controls. The cases included 116 African Americans H.-Q.; Tian, L.; Glessner, J.; Qu, J.; Li, (AA) and 89 European Americans (EA), and the controls included 408 AA and 262 EA. -
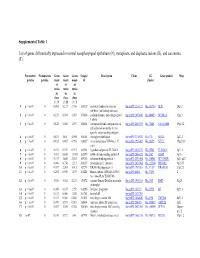
Supplemental Table 1 List of Genes Differentially Expressed In
Supplemental Table 1 List of genes differentially expressed in normal nasopharyngeal epithelium (N), metaplastic and displastic lesions (R), and carcinoma (T). Parametric Permutation Geom Geom Geom Unique Description Clone UG Gene symbol Map p-value p-value mean mean mean id cluster of of of ratios ratios ratios in in in class class class 1 : N 2 : R 3 : T 1 p < 1e-07 0 0.061 0.123 2.708 169329 secretory leukocyte protease IncytePD:2510171 Hs.251754 SLPI 20q12 inhibitor (antileukoproteinase) 2 p < 1e-07 0 0.125 0.394 1.863 163628 sodium channel, nonvoltage-gated IncytePD:1453049 Hs.446415 SCNN1A 12p13 1 alpha 3 p < 1e-07 0 0.122 0.046 1.497 160401 carcinoembryonic antigen-related IncytePD:2060355 Hs.73848 CEACAM6 19q13.2 cell adhesion molecule 6 (non- specific cross reacting antigen) 4 p < 1e-07 0 0.675 1.64 5.594 165101 monoglyceride lipase IncytePD:2174920 Hs.6721 MGLL 3q21.3 5 p < 1e-07 0 0.182 0.487 0.998 166827 nei endonuclease VIII-like 1 (E. IncytePD:1926409 Hs.28355 NEIL1 15q22.33 coli) 6 p < 1e-07 0 0.194 0.339 0.915 162931 hypothetical protein FLJ22418 IncytePD:2816379 Hs.36563 FLJ22418 1p11.1 7 p < 1e-07 0 1.313 0.645 13.593 162399 S100 calcium binding protein P IncytePD:2060823 Hs.2962 S100P 4p16 8 p < 1e-07 0 0.157 1.445 2.563 169315 selenium binding protein 1 IncytePD:2591494 Hs.334841 SELENBP1 1q21-q22 9 p < 1e-07 0 0.046 0.738 1.213 160115 prominin-like 1 (mouse) IncytePD:2070568 Hs.112360 PROML1 4p15.33 10 p < 1e-07 0 0.787 2.264 3.013 167294 HRAS-like suppressor 3 IncytePD:1969263 Hs.37189 HRASLS3 11q12.3 11 p < 1e-07 0 0.292 0.539 1.493 168221 Homo sapiens cDNA FLJ13510 IncytePD:64451 Hs.37896 2 fis, clone PLACE1005146. -

Prenatal Detection of TAR Syndrome in a Fetus with Compound Inheritance of an RBM8A SNP and a 334‑Kb Deletion: a Case Report
MOLECULAR MEDICINE REPORTS 9: 163-165, 2014 Prenatal detection of TAR syndrome in a fetus with compound inheritance of an RBM8A SNP and a 334‑kb deletion: A case report IOANNIS PAPOULIDIS1, EIRINI OIKONOMIDOU1, SANDRO ORRU2, ELISAVET SIOMOU1, MARIA KONTODIOU1, MAKARIOS ELEFTHERIADES3, VASILIOS BACOULAS4, JUAN C. CIGUDOSA5, JAVIER SUELA5, LORETTA THOMAIDIS6 and EMMANOUIL MANOLAKOS1,2 1Laboratory of Genetics, Eurogenetica S.A., Thessaloniki 55133, Greece; 2Department of Medical Genetics, University of Cagliari, Binaghi Hospital, Cagliari I‑09126, Italy; 3Embryocare, Fetal Medicine Unit, Athens 11522; 4Fetal Medicine Centre, Athens 10674, Greece; 5NIMGenetics, Madrid 28049, Spain; 6Department of Pediatrics, Aglaia Kyriakou Children's Hospital, University of Athens, Athens 11527, Greece Received May 30, 2013; Accepted October 16, 2013 DOI: 10.3892/mmr.2013.1788 Abstract. Thrombocytopenia-absent radius syndrome identified the presence of a minimally deleted 200‑kb region (TAR) is a rare genetic disorder that is characterized by the at chromosome band 1q21.1 in patients with TAR, but it is not absence of the radius bone in each forearm and a markedly sufficient to cause the phenotype (3,4). A study identified two reduced platelet count that results in life-threatening bleeding rare single nucleotide polymorphisms (SNPs) in the regulatory episodes (thrombocytopenia). Tar syndrome has been associ- region of the RBM8A gene that are involved in TAR syndrome ated with a deletion of a segment of 1q21.1 cytoband. The through the reduction of the expression of the RBM8A-encoded 1q21.1 deletion syndrome phenotype includes Tar and other Y14 protein (4). The first allele (rs139428292 G>A), which is features such as mental retardation, autism and microcephaly. -

Supplementary Table S1. List of Differentially Expressed
Supplementary table S1. List of differentially expressed transcripts (FDR adjusted p‐value < 0.05 and −1.4 ≤ FC ≥1.4). 1 ID Symbol Entrez Gene Name Adj. p‐Value Log2 FC 214895_s_at ADAM10 ADAM metallopeptidase domain 10 3,11E‐05 −1,400 205997_at ADAM28 ADAM metallopeptidase domain 28 6,57E‐05 −1,400 220606_s_at ADPRM ADP‐ribose/CDP‐alcohol diphosphatase, manganese dependent 6,50E‐06 −1,430 217410_at AGRN agrin 2,34E‐10 1,420 212980_at AHSA2P activator of HSP90 ATPase homolog 2, pseudogene 6,44E‐06 −1,920 219672_at AHSP alpha hemoglobin stabilizing protein 7,27E‐05 2,330 aminoacyl tRNA synthetase complex interacting multifunctional 202541_at AIMP1 4,91E‐06 −1,830 protein 1 210269_s_at AKAP17A A‐kinase anchoring protein 17A 2,64E‐10 −1,560 211560_s_at ALAS2 5ʹ‐aminolevulinate synthase 2 4,28E‐06 3,560 212224_at ALDH1A1 aldehyde dehydrogenase 1 family member A1 8,93E‐04 −1,400 205583_s_at ALG13 ALG13 UDP‐N‐acetylglucosaminyltransferase subunit 9,50E‐07 −1,430 207206_s_at ALOX12 arachidonate 12‐lipoxygenase, 12S type 4,76E‐05 1,630 AMY1C (includes 208498_s_at amylase alpha 1C 3,83E‐05 −1,700 others) 201043_s_at ANP32A acidic nuclear phosphoprotein 32 family member A 5,61E‐09 −1,760 202888_s_at ANPEP alanyl aminopeptidase, membrane 7,40E‐04 −1,600 221013_s_at APOL2 apolipoprotein L2 6,57E‐11 1,600 219094_at ARMC8 armadillo repeat containing 8 3,47E‐08 −1,710 207798_s_at ATXN2L ataxin 2 like 2,16E‐07 −1,410 215990_s_at BCL6 BCL6 transcription repressor 1,74E‐07 −1,700 200776_s_at BZW1 basic leucine zipper and W2 domains 1 1,09E‐06 −1,570 222309_at -

Systematic Elucidation of Neuron-Astrocyte Interaction in Models of Amyotrophic Lateral Sclerosis Using Multi-Modal Integrated Bioinformatics Workflow
ARTICLE https://doi.org/10.1038/s41467-020-19177-y OPEN Systematic elucidation of neuron-astrocyte interaction in models of amyotrophic lateral sclerosis using multi-modal integrated bioinformatics workflow Vartika Mishra et al.# 1234567890():,; Cell-to-cell communications are critical determinants of pathophysiological phenotypes, but methodologies for their systematic elucidation are lacking. Herein, we propose an approach for the Systematic Elucidation and Assessment of Regulatory Cell-to-cell Interaction Net- works (SEARCHIN) to identify ligand-mediated interactions between distinct cellular com- partments. To test this approach, we selected a model of amyotrophic lateral sclerosis (ALS), in which astrocytes expressing mutant superoxide dismutase-1 (mutSOD1) kill wild-type motor neurons (MNs) by an unknown mechanism. Our integrative analysis that combines proteomics and regulatory network analysis infers the interaction between astrocyte-released amyloid precursor protein (APP) and death receptor-6 (DR6) on MNs as the top predicted ligand-receptor pair. The inferred deleterious role of APP and DR6 is confirmed in vitro in models of ALS. Moreover, the DR6 knockdown in MNs of transgenic mutSOD1 mice attenuates the ALS-like phenotype. Our results support the usefulness of integrative, systems biology approach to gain insights into complex neurobiological disease processes as in ALS and posit that the proposed methodology is not restricted to this biological context and could be used in a variety of other non-cell-autonomous communication -

The Genetic Architecture of Fasting Plasma Triglyceride Response to Fenofibrate Treatment
European Journal of Human Genetics (2008) 16, 603–613 & 2008 Nature Publishing Group All rights reserved 1018-4813/08 $30.00 www.nature.com/ejhg ARTICLE The genetic architecture of fasting plasma triglyceride response to fenofibrate treatment Jennifer A Smith*,1, Donna K Arnett2, Reagan J Kelly1, Jose M Ordovas3, Yan V Sun1, Paul N Hopkins4,JamesEHixson5,RobertJStraka6, James M Peacock7 and Sharon LR Kardia1 1Department of Epidemiology, School of Public Health, University of Michigan, Ann Arbor, MI, USA; 2Department of Epidemiology, School of Public Health, University of Alabama at Birmingham, Birmingham, AL, USA; 3Nutrition and Genomics Laboratory, Jean Mayer-United States Department of Agriculture Human Nutrition Research Center on Aging, Tufts University, Boston, MA, USA; 4Cardiovascular Genetics Research, University of Utah, Salt Lake City, UT, USA; 5Human Genetics Center, University of Texas Health Science, Houston, TX, USA; 6Department of Experimental and Clinical Pharmacology, College of Pharmacy, University of Minnesota, Minneapolis, MN, USA; 7Department of Epidemiology, School of Public Health, University of Minnesota, Minneapolis, MN, USA Metabolic response to the triglyceride (TG)-lowering drug, fenofibrate, is shaped by interactions between genetic and environmental factors, yet knowledge regarding the genetic determinants of this response is primarily limited to single-gene effects. Since very low-density lipoprotein (VLDL) is the central carrier of fasting TG, identifying factors that affect both total TG and VLDL–TG response to fenofibrate is critical for predicting individual fenofibrate response. As part of the Genetics of Lipid Lowering Drugs and Diet Network (GOLDN) study, 688 individuals from 161 families were genotyped for 91 single-nucleotide polymorphisms (SNPs) in 25 genes known to be involved in lipoprotein metabolism. -

Comprehensive Analysis Reveals Novel Gene Signature in Head and Neck Squamous Cell Carcinoma: Predicting Is Associated with Poor Prognosis in Patients
5892 Original Article Comprehensive analysis reveals novel gene signature in head and neck squamous cell carcinoma: predicting is associated with poor prognosis in patients Yixin Sun1,2#, Quan Zhang1,2#, Lanlin Yao2#, Shuai Wang3, Zhiming Zhang1,2 1Department of Breast Surgery, The First Affiliated Hospital of Xiamen University, School of Medicine, Xiamen University, Xiamen, China; 2School of Medicine, Xiamen University, Xiamen, China; 3State Key Laboratory of Cellular Stress Biology, School of Life Sciences, Xiamen University, Xiamen, China Contributions: (I) Conception and design: Y Sun, Q Zhang; (II) Administrative support: Z Zhang; (III) Provision of study materials or patients: Y Sun, Q Zhang; (IV) Collection and assembly of data: Y Sun, L Yao; (V) Data analysis and interpretation: Y Sun, S Wang; (VI) Manuscript writing: All authors; (VII) Final approval of manuscript: All authors. #These authors contributed equally to this work. Correspondence to: Zhiming Zhang. Department of Surgery, The First Affiliated Hospital of Xiamen University, Xiamen, China. Email: [email protected]. Background: Head and neck squamous cell carcinoma (HNSC) remains an important public health problem, with classic risk factors being smoking and excessive alcohol consumption and usually has a poor prognosis. Therefore, it is important to explore the underlying mechanisms of tumorigenesis and screen the genes and pathways identified from such studies and their role in pathogenesis. The purpose of this study was to identify genes or signal pathways associated with the development of HNSC. Methods: In this study, we downloaded gene expression profiles of GSE53819 from the Gene Expression Omnibus (GEO) database, including 18 HNSC tissues and 18 normal tissues. -

Identifying Rare Genetic Variation in Obsessive-Compulsive Disorder
Yale University EliScholar – A Digital Platform for Scholarly Publishing at Yale Yale Medicine Thesis Digital Library School of Medicine January 2020 Identifying Rare Genetic Variation In Obsessive-Compulsive Disorder Sarah Abdallah Follow this and additional works at: https://elischolar.library.yale.edu/ymtdl Recommended Citation Abdallah, Sarah, "Identifying Rare Genetic Variation In Obsessive-Compulsive Disorder" (2020). Yale Medicine Thesis Digital Library. 3876. https://elischolar.library.yale.edu/ymtdl/3876 This Open Access Thesis is brought to you for free and open access by the School of Medicine at EliScholar – A Digital Platform for Scholarly Publishing at Yale. It has been accepted for inclusion in Yale Medicine Thesis Digital Library by an authorized administrator of EliScholar – A Digital Platform for Scholarly Publishing at Yale. For more information, please contact [email protected]. Identifying Rare Genetic Variation in Obsessive-Compulsive Disorder A Thesis Submitted to the Yale University School of Medicine in Partial Fulfillment of the Requirements for the Degree of Doctor of Medicine by Sarah Barbara Abdallah 2020 ABSTRACT IDENTIFYING RARE GENETIC VARIATION IN OBSESSIVE-COMPULSIVE DISORDER Sarah B. Abdallah, Carolina Cappi, Emily Olfson, and Thomas V. Fernandez. Child Study Center, Yale University School of Medicine, New Haven, CT Obsessive-compulsive disorder (OCD) is a neuropsychiatric developmental disorder with known heritability (estimates ranging from 27%-80%) but poorly understood etiology. Current treatments are not fully effective in addressing chronic functional impairments and distress caused by the disorder, providing an impetus to study the genetic basis of OCD in the hopes of identifying new therapeutic targets. We previously demonstrated a significant contribution to OCD risk from likely damaging de novo germline DNA sequence variants, which arise spontaneously in the parental germ cells or zygote instead of being inherited from a parent, and we successfully used these identified variants to implicate new OCD risk genes. -

Common Variant of PDZ Domain Containing 1 (PDZK1) Gene Is Associated with Gout Susceptibility: a Replication Study and Meta-Analysis in Japanese Population
Drug Metabolism and Pharmacokinetics xxx (2016) 1e3 Contents lists available at ScienceDirect Drug Metabolism and Pharmacokinetics journal homepage: http://www.journals.elsevier.com/drug-metabolism-and- pharmacokinetics Note Common variant of PDZ domain containing 1 (PDZK1) gene is associated with gout susceptibility: A replication study and meta-analysis in Japanese population * Toshihide Higashino a, 1, Hirotaka Matsuo a, , 1, Masayuki Sakiyama a, Akiyoshi Nakayama a, Takahiro Nakamura b, Tappei Takada c, Hiraku Ogata a, Yusuke Kawamura a, Makoto Kawaguchi a, Mariko Naito d, Sayo Kawai d, Yuzo Takada e, Hiroshi Ooyama f, Hiroshi Suzuki c, Nariyoshi Shinomiya a a Department of Integrative Physiology and Bio-Nano Medicine, National Defense Medical College, Tokorozawa, Japan b Laboratory of Mathematics, National Defense Medical College, Tokorozawa, Japan c Department of Pharmacy, The University of Tokyo Hospital, Faculty of Medicine, The University of Tokyo, Tokyo, Japan d Department of Preventive Medicine, Nagoya University Graduate School of Medicine, Nagoya, Japan e Faculty of Medical Science, Teikyo University of Science, Tokyo, Japan f Ryougoku East Gate Clinic, Tokyo, Japan article info abstract Article history: PDZ domain containing 1 (PDZK1) is a scaffold protein that organizes a transportsome and regulates Received 24 May 2016 several transporters' functions including urate and drug transporters. Therefore, PDZK1 in renal proximal Received in revised form tubules may affect serum uric acid levels through PDZK1-binding renal urate transporters. Two previous 4 July 2016 studies in Japanese male population reported that a PDZK1 single nucleotide polymorphism (SNP), Accepted 25 July 2016 rs12129861, was not associated with gout. In the present study, we performed a further association Available online xxx analysis between gout and rs12129861 in a different large-scale Japanese male population and a meta- analysis with previous Japanese population studies.