The LMO2 Oncogene Regulates DNA Replication in Hematopoietic Cells
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Characterization of a Pediatric T-Cell Acute Lymphoblastic Leukemia Patient with Simultaneous LYL1 and LMO2 Rearrangements
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Erasmus University Digital Repository Articles and Brief Reports Acute Lymphoblastic Leukemia Characterization of a pediatric T-cell acute lymphoblastic leukemia patient with simultaneous LYL1 and LMO2 rearrangements Irene Homminga, 1 Maartje J. Vuerhard, 1 Anton W. Langerak, 2 Jessica Buijs-Gladdines, 1 Rob Pieters, 1 and Jules P.P. Meijerink 1 1Department of Pediatric Oncology/Hematology, Erasmus MC/Sophia Children’s Hospital, Rotterdam; and 2Department of Immunology, Erasmus MC, Rotterdam, The Netherlands ABSTRACT Translocation of the LYL1 oncogene are rare in T-cell acute consistently clustered along with cases having TAL1 or lymphoblastic leukemia, whereas the homologous TAL1 LMO2 rearrangements. Therefore, LYL1 -rearranged cases are gene is rearranged in approximately 20% of patients. not necessarily associated with immature T-cell develop - Previous gene-expression studies have identified an imma - ment, despite high LYL1 levels, but elicit a TALLMO expres - ture T-cell acute lymphoblastic leukemia subgroup with high sion signature. LYL1 expression in the absence of chromosomal aberrations. Molecular characterization of a t(7;19)(q34;p13) in a pediatric Key words: T-ALL, pediatric, LYL1, LMO2, rearrangements. T-cell acute lymphoblastic leukemia patient led to the identi - fication of a translocation between the TRB@ and LYL1 loci. Citation: Homminga I, Vuerhard MJ, Langerak AW, Buijs- Similar to incidental T-cell acute lymphoblastic leukemia Gladdines J, Pieters R, and Meijerink JPP. Characterization of a cases with synergistic, double translocations affecting pediatric T-cell acute lymphoblastic leukemia patient with simul - TAL1/2 and LMO1/2 oncogenes, this LYL1 -translocated taneous LYL1 and LMO2 rearrangements. -

Molecular Profile of Tumor-Specific CD8+ T Cell Hypofunction in a Transplantable Murine Cancer Model
Downloaded from http://www.jimmunol.org/ by guest on September 25, 2021 T + is online at: average * The Journal of Immunology , 34 of which you can access for free at: 2016; 197:1477-1488; Prepublished online 1 July from submission to initial decision 4 weeks from acceptance to publication 2016; doi: 10.4049/jimmunol.1600589 http://www.jimmunol.org/content/197/4/1477 Molecular Profile of Tumor-Specific CD8 Cell Hypofunction in a Transplantable Murine Cancer Model Katherine A. Waugh, Sonia M. Leach, Brandon L. Moore, Tullia C. Bruno, Jonathan D. Buhrman and Jill E. Slansky J Immunol cites 95 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html http://www.jimmunol.org/content/suppl/2016/07/01/jimmunol.160058 9.DCSupplemental This article http://www.jimmunol.org/content/197/4/1477.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2016 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 25, 2021. The Journal of Immunology Molecular Profile of Tumor-Specific CD8+ T Cell Hypofunction in a Transplantable Murine Cancer Model Katherine A. -

DNA·RNA Triple Helix Formation Can Function As a Cis-Acting Regulatory
DNA·RNA triple helix formation can function as a cis-acting regulatory mechanism at the human β-globin locus Zhuo Zhoua, Keith E. Gilesa,b,c, and Gary Felsenfelda,1 aLaboratory of Molecular Biology, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, MD 20892; bUniversity of Alabama at Birmingham Stem Cell Institute, University of Alabama at Birmingham, Birmingham, AL 35294; and cDepartment of Biochemistry and Molecular Genetics, University of Alabama at Birmingham, Birmingham, AL 35294 Contributed by Gary Felsenfeld, February 4, 2019 (sent for review January 4, 2019; reviewed by James Douglas Engel and Sergei M. Mirkin) We have identified regulatory mechanisms in which an RNA tran- of the criteria necessary to establish the presence of a triplex script forms a DNA duplex·RNA triple helix with a gene or one of its structure, we first describe and characterize triplex formation at regulatory elements, suggesting potential auto-regulatory mecha- the FAU gene in human erythroid K562 cells. FAU encodes a nisms in vivo. We describe an interaction at the human β-globin protein that is a fusion containing fubi, a ubiquitin-like protein, locus, in which an RNA segment embedded in the second intron of and ribosomal protein S30. Although fubi function is unknown, the β-globin gene forms a DNA·RNA triplex with the HS2 sequence posttranslational processing produces S30, a component of the within the β-globin locus control region, a major regulator of glo- 40S ribosome. We used this system to refine methods necessary bin expression. We show in human K562 cells that the triplex is to detect triplex formation and to distinguish it from R-loop stable in vivo. -

Iron Depletion Reduces Abce1 Transcripts While Inducing The
Preprints (www.preprints.org) | NOT PEER-REVIEWED | Posted: 22 October 2019 doi:10.20944/preprints201910.0252.v1 1 Research Article 2 Iron depletion Reduces Abce1 Transcripts While 3 Inducing the Mitophagy Factors Pink1 and Parkin 4 Jana Key 1,2, Nesli Ece Sen 1, Aleksandar Arsovic 1, Stella Krämer 1, Robert Hülse 1, Suzana 5 Gispert-Sanchez 1 and Georg Auburger 1,* 6 1 Experimental Neurology, Goethe University Medical School, 60590 Frankfurt am Main; 7 2 Faculty of Biosciences, Goethe-University Frankfurt am Main, Germany 8 * Correspondence: [email protected] 9 10 Abstract: Lifespan extension was recently achieved in Caenorhabditis elegans nematodes by 11 mitochondrial stress and mitophagy, triggered via iron depletion. Conversely in man, deficient 12 mitophagy due to Pink1/Parkin mutations triggers iron accumulation in patient brain and limits 13 survival. We now aimed to identify murine fibroblast factors, which adapt their mRNA expression 14 to acute iron manipulation, relate to mitochondrial dysfunction and may influence survival. After 15 iron depletion, expression of the plasma membrane receptor Tfrc with its activator Ireb2, the 16 mitochondrial membrane transporter Abcb10, the heme-release factor Pgrmc1, the heme- 17 degradation enzyme Hmox1, the heme-binding cholesterol metabolizer Cyp46a1, as well as the 18 mitophagy regulators Pink1 and Parkin showed a negative correlation to iron levels. After iron 19 overload, these factors did not change expression. Conversely, a positive correlation of mRNA levels 20 with both conditions of iron availability was observed for the endosomal factors Slc11a2 and Steap2, 21 as well as for the iron-sulfur-cluster (ISC)-containing factors Ppat, Bdh2 and Nthl1. -

Ldb1 Is Essential for Development of Nkx2.1 Lineage Derived Gabaergic and Cholinergic Neurons in the Telencephalon
Developmental Biology 385 (2014) 94–106 Contents lists available at ScienceDirect Developmental Biology journal homepage: www.elsevier.com/locate/developmentalbiology Ldb1 is essential for development of Nkx2.1 lineage derived GABAergic and cholinergic neurons in the telencephalon Yangu Zhao a,n,1, Pierre Flandin b,2, Daniel Vogt b, Alexander Blood a, Edit Hermesz a,3, Heiner Westphal a, John L. R. Rubenstein b,nn a Program on Genomics of Differentiation, Eunice Kennedy Shriver National Institute of Child Health and Human Development, NIH, Bethesda, MD 20892, USA b Department of Psychiatry and the Nina Ireland Laboratory of Developmental Neurobiology, University of California San Francisco, San Francisco, CA 94143, USA article info abstract Article history: The progenitor zones of the embryonic mouse ventral telencephalon give rise to GABAergic and cholinergic Received 1 June 2013 neurons. We have shown previously that two LIM-homeodomain (LIM-HD) transcription factors, Lhx6 and Received in revised form Lhx8, that are downstream of Nkx2.1, are critical for the development of telencephalic GABAergic and 8 October 2013 cholinergic neurons. Here we investigate the role of Ldb1, a nuclear protein that binds directly to all LIM-HD Accepted 9 October 2013 factors, in the development of these ventral telencephalon derived neurons. We show that Ldb1 is expressed Available online 21 October 2013 in the Nkx2.1 cell lineage during embryonic development and in mature neurons. Conditional deletion of Ldb1 Keywords: causes defects in the expression of a series of genes in the ventral telencephalon and severe impairment in the Differentiation tangential migration of cortical interneurons from the ventral telencephalon. -

Open Dogan Phdthesis Final.Pdf
The Pennsylvania State University The Graduate School Eberly College of Science ELUCIDATING BIOLOGICAL FUNCTION OF GENOMIC DNA WITH ROBUST SIGNALS OF BIOCHEMICAL ACTIVITY: INTEGRATIVE GENOME-WIDE STUDIES OF ENHANCERS A Dissertation in Biochemistry, Microbiology and Molecular Biology by Nergiz Dogan © 2014 Nergiz Dogan Submitted in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy August 2014 ii The dissertation of Nergiz Dogan was reviewed and approved* by the following: Ross C. Hardison T. Ming Chu Professor of Biochemistry and Molecular Biology Dissertation Advisor Chair of Committee David S. Gilmour Professor of Molecular and Cell Biology Anton Nekrutenko Professor of Biochemistry and Molecular Biology Robert F. Paulson Professor of Veterinary and Biomedical Sciences Philip Reno Assistant Professor of Antropology Scott B. Selleck Professor and Head of the Department of Biochemistry and Molecular Biology *Signatures are on file in the Graduate School iii ABSTRACT Genome-wide measurements of epigenetic features such as histone modifications, occupancy by transcription factors and coactivators provide the opportunity to understand more globally how genes are regulated. While much effort is being put into integrating the marks from various combinations of features, the contribution of each feature to accuracy of enhancer prediction is not known. We began with predictions of 4,915 candidate erythroid enhancers based on genomic occupancy by TAL1, a key hematopoietic transcription factor that is strongly associated with gene induction in erythroid cells. Seventy of these DNA segments occupied by TAL1 (TAL1 OSs) were tested by transient transfections of cultured hematopoietic cells, and 56% of these were active as enhancers. Sixty-six TAL1 OSs were evaluated in transgenic mouse embryos, and 65% of these were active enhancers in various tissues. -
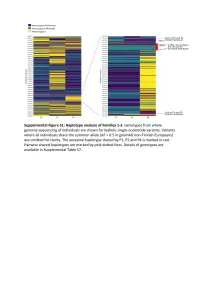
Supplementary Text and Figures.Pdf
Supplemental Figure S2. PRIM1 immunoblots (20-75 kDa). (A) Corresponds to Figure 2D, with additional bands seen in P2 at 75 kDa and 25 kDa. (B) Independent experiment with cell lysates from C and P2 in which additional bands at 25 kDa not evident. (C) Corresponds to Figure 2D. No additional band visible at 25 kDa. (D) Validation of monoclonal antibody (8G10) with siRNA to PRIM1 in HeLa cells. siLUC, luciferase negative control. siPRIM1, siRNA targeting PRIM1 transcript. Vinculin used as a loading control. Supplemental Figure S3: Predicted destabilisation of PRIM1 by the C301R substitution. (A) Cysteine 301 lies in a buried hydrophobic region in PRIM1. DNA primase dimer crystal structure (PDB: 4BPU). PRIM1 residues shaded according to solvent accessibility. Cysteine 301, substituted to arginine in P5, lies in a buried hydrophobic region. (B) Cys301 is evolutionarily conserved in vertebrates. Neither arginine or other large or charged amino acids are observed at this position in other species. (C) The C301R substitution observed in P5 is predicted to lead to destabilisation of all available PRIM1 crystal structures. In contrast, substitution with leucine or threonine, as observed in some orthologous proteins, is not predicted to lead to destabilisation. Data points, predicted changes in free-energy (ΔΔG) from FoldX plotted for each of 14 available crystal structures. (D) Immunoblotting of RPE1 cells transfected with dual reporter constructs (as described in Figure 3B and Materials and Methods) confirms production of PRIM1-GFP and FLAG-SR at expected molecular weights and shows reduced protein levels for the C301R variant. n=2 experiments shown. C301R protein levels relative to WT after normalization to actin loading control indicated underneath the blot. -

The Role of Noncoding Mutations in Blood Cancers Sunniyat Rahman and Marc R
© 2019. Published by The Company of Biologists Ltd | Disease Models & Mechanisms (2019) 12, dmm041988. doi:10.1242/dmm.041988 REVIEW The role of noncoding mutations in blood cancers Sunniyat Rahman and Marc R. Mansour* ABSTRACT survival in adult acute myeloid leukaemia (AML) and ALL are The search for oncogenic mutations in haematological malignancies still less than 50%, with significant treatment challenges including has largely focused on coding sequence variants. These variants treatment-resistant disease, clonal heterogeneity of the underlying have been critical in understanding these complex cancers in greater disease, treatment-associated toxicities and poor tolerance for detail, ultimately leading to better disease monitoring, subtyping and intensive treatment regimens, particularly in older patients with prognostication. In contrast, the search for oncogenic variants in the comorbidities (Kansagra et al., 2018). Because of this, there is a noncoding genome has proven to be challenging given the vastness significant impetus to discover new genetic aberrations, including of the search space, the intrinsic difficulty in assessing the impact of driver mutations (see Box 1 for a glossary of terms), that may variants that do not code for functional proteins, and our still primitive provide insight into the mechanisms of malignant haematopoiesis, understanding of the function harboured by large parts of the as well as offering novel therapeutic opportunities. noncoding genome. Recent studies have broken ground on this Detailed genetic characterisation of haematological malignancies quest, identifying somatically acquired and recurrent mutations in the has already identified alterations that are now being used for better noncoding genome that activate the expression of proto-oncogenes. diagnosis, prognostication, subtype identification and to inform In this Review, we explore some of the best-characterised examples therapeutic decisions (Taylor et al., 2017). -

Cell Growth-Regulated Expression of Mammalian MCM5 and MCM6 Genes Mediated by the Transcription Factor E2F
Oncogene (1999) 18, 2299 ± 2309 ã 1999 Stockton Press All rights reserved 0950 ± 9232/99 $12.00 http://www.stockton-press.co.uk/onc Cell growth-regulated expression of mammalian MCM5 and MCM6 genes mediated by the transcription factor E2F Kiyoshi Ohtani1, Ritsuko Iwanaga1, Masataka Nakamura*,1, Masa-aki Ikeda2, Norikazu Yabuta3, Hiromichi Tsuruga3 and Hiroshi Nojima3 1Human Gene Sciences Center, Tokyo Medical and Dental University, Tokyo 113-8510, Japan 2Department of Developmental Biology, Graduate School of Dentistry, Tokyo Medical and Dental University, Tokyo 113-8549, Japan; 3Department of Molecular Genetics, Research Institute for Microbial Diseases, Osaka University, Suita 565-0871, Japan Initiation of DNA replication requires the function of family (MCM2-7) that have been identi®ed in yeast, MCM gene products, which participate in ensuring that Xenopus, and human. Mcm proteins seem to regulate DNA replication occurs only once in the cell cycle. the initiation at the replication origin where the loading Expression of all mammalian genes of the MCM family of the proteins onto the origin recognition complex is induced by growth stimulation, unlike yeast, and the (ORC) is regulated by Cdc6 and cyclin-dependent mRNA levels peak at G1/S boundary. In this study, we kinases (Donovan et al., 1997; Tanaka et al., 1997). examined the transcriptional activities of isolated human However, the mechanism(s) by which Mcm proteins MCM gene promoters. Human MCM5 and MCM6 control the initiation of DNA replication remains promoters with mutation in the E2F sites failed in unclear. promoter regulation following serum stimulation and Xenopus Mcm proteins seem to be able to access exogenous E2F expression. -

Supplementary Table S5. Differentially Expressed Gene Lists of PD-1High CD39+ CD8 Tils According to 4-1BB Expression Compared to PD-1+ CD39- CD8 Tils
BMJ Publishing Group Limited (BMJ) disclaims all liability and responsibility arising from any reliance Supplemental material placed on this supplemental material which has been supplied by the author(s) J Immunother Cancer Supplementary Table S5. Differentially expressed gene lists of PD-1high CD39+ CD8 TILs according to 4-1BB expression compared to PD-1+ CD39- CD8 TILs Up- or down- regulated genes in Up- or down- regulated genes Up- or down- regulated genes only PD-1high CD39+ CD8 TILs only in 4-1BBneg PD-1high CD39+ in 4-1BBpos PD-1high CD39+ CD8 compared to PD-1+ CD39- CD8 CD8 TILs compared to PD-1+ TILs compared to PD-1+ CD39- TILs CD39- CD8 TILs CD8 TILs IL7R KLRG1 TNFSF4 ENTPD1 DHRS3 LEF1 ITGA5 MKI67 PZP KLF3 RYR2 SIK1B ANK3 LYST PPP1R3B ETV1 ADAM28 H2AC13 CCR7 GFOD1 RASGRP2 ITGAX MAST4 RAD51AP1 MYO1E CLCF1 NEBL S1PR5 VCL MPP7 MS4A6A PHLDB1 GFPT2 TNF RPL3 SPRY4 VCAM1 B4GALT5 TIPARP TNS3 PDCD1 POLQ AKAP5 IL6ST LY9 PLXND1 PLEKHA1 NEU1 DGKH SPRY2 PLEKHG3 IKZF4 MTX3 PARK7 ATP8B4 SYT11 PTGER4 SORL1 RAB11FIP5 BRCA1 MAP4K3 NCR1 CCR4 S1PR1 PDE8A IFIT2 EPHA4 ARHGEF12 PAICS PELI2 LAT2 GPRASP1 TTN RPLP0 IL4I1 AUTS2 RPS3 CDCA3 NHS LONRF2 CDC42EP3 SLCO3A1 RRM2 ADAMTSL4 INPP5F ARHGAP31 ESCO2 ADRB2 CSF1 WDHD1 GOLIM4 CDK5RAP1 CD69 GLUL HJURP SHC4 GNLY TTC9 HELLS DPP4 IL23A PITPNC1 TOX ARHGEF9 EXO1 SLC4A4 CKAP4 CARMIL3 NHSL2 DZIP3 GINS1 FUT8 UBASH3B CDCA5 PDE7B SOGA1 CDC45 NR3C2 TRIB1 KIF14 TRAF5 LIMS1 PPP1R2C TNFRSF9 KLRC2 POLA1 CD80 ATP10D CDCA8 SETD7 IER2 PATL2 CCDC141 CD84 HSPA6 CYB561 MPHOSPH9 CLSPN KLRC1 PTMS SCML4 ZBTB10 CCL3 CA5B PIP5K1B WNT9A CCNH GEM IL18RAP GGH SARDH B3GNT7 C13orf46 SBF2 IKZF3 ZMAT1 TCF7 NECTIN1 H3C7 FOS PAG1 HECA SLC4A10 SLC35G2 PER1 P2RY1 NFKBIA WDR76 PLAUR KDM1A H1-5 TSHZ2 FAM102B HMMR GPR132 CCRL2 PARP8 A2M ST8SIA1 NUF2 IL5RA RBPMS UBE2T USP53 EEF1A1 PLAC8 LGR6 TMEM123 NEK2 SNAP47 PTGIS SH2B3 P2RY8 S100PBP PLEKHA7 CLNK CRIM1 MGAT5 YBX3 TP53INP1 DTL CFH FEZ1 MYB FRMD4B TSPAN5 STIL ITGA2 GOLGA6L10 MYBL2 AHI1 CAND2 GZMB RBPJ PELI1 HSPA1B KCNK5 GOLGA6L9 TICRR TPRG1 UBE2C AURKA Leem G, et al. -

Congenital Microcephaly
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Sussex Research Online American Journal of Medical Genetics Part C (Seminars in Medical Genetics) ARTICLE Congenital Microcephaly DIANA ALCANTARA AND MARK O'DRISCOLL* The underlying etiologies of genetic congenital microcephaly are complex and multifactorial. Recently, with the exponential growth in the identification and characterization of novel genetic causes of congenital microcephaly, there has been a consolidation and emergence of certain themes concerning underlying pathomechanisms. These include abnormal mitotic microtubule spindle structure, numerical and structural abnormalities of the centrosome, altered cilia function, impaired DNA repair, DNA Damage Response signaling and DNA replication, along with attenuated cell cycle checkpoint proficiency. Many of these processes are highly interconnected. Interestingly, a defect in a gene whose encoded protein has a canonical function in one of these processes can often have multiple impacts at the cellular level involving several of these pathways. Here, we overview the key pathomechanistic themes underlying profound congenital microcephaly, and emphasize their interconnected nature. © 2014 Wiley Periodicals, Inc. KEY WORDS: cell division; mitosis; DNA replication; cilia How to cite this article: Alcantara D, O'Driscoll M. 2014. Congenital microcephaly. Am J Med Genet Part C Semin Med Genet 9999:1–16. INTRODUCTION mid‐gestation although glial cell division formation of the various cortical layers. and consequent brain volume enlarge- Furthermore, differentiating and devel- Congenital microcephaly, an occipital‐ ment does continue after birth [Spalding oping neurons must migrate to their frontal circumference of equal to or less et al., 2005]. Impaired neurogenesis is defined locations to construct the com- than 2–3 standard deviations below the therefore most obviously reflected clini- plex architecture and laminar layered age‐related population mean, denotes cally as congenital microcephaly. -

LMO T-Cell Translocation Oncogenes Typify Genes Activated by Chromosomal Translocations That Alter Transcription and Developmental Processes
Downloaded from genesdev.cshlp.org on September 25, 2021 - Published by Cold Spring Harbor Laboratory Press PERSPECTIVE LMO T-cell translocation oncogenes typify genes activated by chromosomal translocations that alter transcription and developmental processes Terence H. Rabbitts1 Medical Research Council (MRC) Laboratory of Molecular Biology, Division of Protein and Nucleic Acid Chemistry, Cambridge CB2 2QH, UK The cytogenetic analysis of tumors, particularly those of ment (immunoglobulin or T-cell receptor) occurs and oc- hematopoietic origin, has revealed that reciprocal chro- casionally mediates chromosomal translocation. This mosomal translocations are recurring features of these type of translocation causes oncogene activation result- tumors. Further from the initial recognition of the trans- ing from the new chromosomal environment of the re- location t(9;22) (Nowell and Hungerford 1960; Rowley arranged gene. In general, this means inappropriate gene 1973), it has become clear that particular chromosomal expression. The second, and probably the most common translocations are found consistently in specific tumor outcome of chromosomal translocations, is gene fusion subtypes. A principle example of this is the translocation in which exons from a gene on each of the involved chro- t(8;14)(q24;q32.1) invariably found in the human B cell mosomes are linked after the chromosomal transloca- tumor Burkitt’s lymphoma (Manolov and Manolov 1972; tion, resulting in a fusion mRNA and protein. This type Zech et al. 1976). The link with the immunoglobulin of event is found in many cases of hematopoietic tumors H-chain locus on chromosome 14, band q32.1 (Croce et and in the sarcomas. al. 1979; Hobart et al.