Investigation of Plants Containing Unusual Ecdysteroids with Ring in Their Side-Chain
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
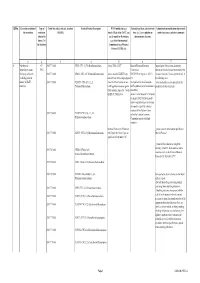
Qrno. 1 2 3 4 5 6 7 1 CP 2903 77 100 0 Cfcl3
QRNo. General description of Type of Tariff line code(s) affected, based on Detailed Product Description WTO Justification (e.g. National legal basis and entry into Administration, modification of previously the restriction restriction HS(2012) Article XX(g) of the GATT, etc.) force (i.e. Law, regulation or notified measures, and other comments (Symbol in and Grounds for Restriction, administrative decision) Annex 2 of e.g., Other International the Decision) Commitments (e.g. Montreal Protocol, CITES, etc) 12 3 4 5 6 7 1 Prohibition to CP 2903 77 100 0 CFCl3 (CFC-11) Trichlorofluoromethane Article XX(h) GATT Board of Eurasian Economic Import/export of these ozone destroying import/export ozone CP-X Commission substances from/to the customs territory of the destroying substances 2903 77 200 0 CF2Cl2 (CFC-12) Dichlorodifluoromethane Article 46 of the EAEU Treaty DECISION on August 16, 2012 N Eurasian Economic Union is permitted only in (excluding goods in dated 29 may 2014 and paragraphs 134 the following cases: transit) (all EAEU 2903 77 300 0 C2F3Cl3 (CFC-113) 1,1,2- 4 and 37 of the Protocol on non- On legal acts in the field of non- _to be used solely as a raw material for the countries) Trichlorotrifluoroethane tariff regulation measures against tariff regulation (as last amended at 2 production of other chemicals; third countries Annex No. 7 to the June 2016) EAEU of 29 May 2014 Annex 1 to the Decision N 134 dated 16 August 2012 Unit list of goods subject to prohibitions or restrictions on import or export by countries- members of the -

Quality Assessment of Rhaponticum Carthamoides (Willd.) Iljin As
Timofeev N.P., Lapin A.A., Zelenkov V.N. Quality Assessment of Rhaponticum carthamoides (Willd.) Iljin as Medicinal Raw Material by the Bromic Antioxidant Capacity Estimation // Journal Chemistry and Computational Simulation: Butlerov Communications, 2006, 8(2): 35-40. QUALITY ASSESSMENT OF RHAPONTICUM CARTHAMOIDES (WILLD.) ILJIN AS MEDICINAL RAW MATERIAL BY THE BROMIC ANTIOXIDANT CAPACITY ESTIMATION N.P. Timofeev1, A.A. Lapin2, V.N. Zelenkov3 1 Collective farm BIO, 165650, Russia, Koryazhma; e-mail: [email protected] 2 Arbuzov’s Institute of organic and physical chemistry, KazSC of the Russian Academy of Sci- ence, Kazan; 3 Russian academy of natural sciences, Moscow ABSCTRACT The possibility of an express assessment of quality medicinal raw materials Rhaponticum carthamoides (Willd.) Iljin is investigated, at various stages of harvesting and storage by method Bromic Antioxidant Capacity Estimation (BACE), after extraction of sample in an aqueous solu- tion. Revealed that phytoecdysteroids R. carthamoides extracted in aqueous solution at level of ethanol, have the temperature resistance. Among the investigated 17 species of industrial medici- nal plants value R. carthamoides has the greatest value BACE, exceeding values of other species from 2-3 up to 5-12 times. Change BACE of leaf organs during the growing season was correlated with the dynamics of the concentration ecdysteroid 20-hydroxyecdysone, as defined by method of the reversed-phase highly effective liquid chromatography (RP-HPLC). Higher values for both indicators were also true for the early phases of vegetation. Established a connection between the loss of active sub- stances and reduction value BACE during storage. On the safety of 20-hydroxyecdysone and the value BACE strongly influenced by the pres- ence of impurities in raw materials, infected microflora. -

Szent István University Faculty of Horticultural Science Department of Genetics and Plant Breeding
10.14751/SZIE.2016.071 SZENT ISTVÁN UNIVERSITY FACULTY OF HORTICULTURAL SCIENCE DEPARTMENT OF GENETICS AND PLANT BREEDING ANALYSIS OF THE GLYCOSIDE BIOSYNTHESIS IN RHODIOLA ROSEA L. DOCTORAL (Ph.D.) DISSERTATION SEYED IMAN MIRMAZLOUM SUPERVISOR: BENYÓNÉ DR. GYÖRGY ZSUZSANNA BUDAPEST 2016 1 10.14751/SZIE.2016.071 Ph.D School Name: Doctoral School of Horticultural Science Field: Crop Sciences and Horticulture Head of the Ph.D school: Prof. Dr. Zámboriné Németh Éva, Doctor of the Hungarian Academy of Science Head of Department of Medicinal and Aromatic SZENT ISTVÁN UNIVERSITY, Faculty of Horticultural Science Supervisor: Benyóné Dr. György Zsuzsanna Department of Genetics and Plant Breeding SZENT ISTVÁN UNIVERSITY, Faculty of Horticultural Sciences The applicant met the requirement of the Ph.D regulations of the SZENT ISTVÁN UNIVERSITY and the thesis is accepted for the defense process. .................................. ................................ Head of Ph.D. School Supervisor 2 10.14751/SZIE.2016.071 JURY MEMBERS: Chairman: Dr. Jenő Bernáth DSc Scientific committee: Dr. István Papp DSc Dr. György Bisztray PhD Dr. Éva Szőke DSc Dr. Alexandra Soltész PhD Opponents: Dr. Tamás Deák PhD Dr. Ágnes Dalmadi PhD 3 10.14751/SZIE.2016.071 CONTENT LIST OF ABBREVIATIONS…………………………………………………………………….....6 1. INTRODUCTION AND OBJECTIVES ....................................................................................... 7 2. LITERATURE REVIEW ........................................................................................................ -

Effect of Climate on Plant Growth and Level of Adaptogenic Compounds
® The European Journal of Plant Science and Biotechnology ©2011 Global Science Books Effect of Climate on Plant Growth and Level of Adaptogenic Compounds in Maral Root (Leuzea charthamoides (Willd.) DC.), Crowned Saw-wort (Serratula coronata L.) and Roseroot (Rhodiola rosea L.) Inger Martinussen1* • Vladimir Volodin2 • Svetlana Volodina2 • Eivind Uleberg1 1 Norwegian Institute for Agricultural and Environmental Research, Arctic Agriculture and Land Use Division, Box 2284, N-9269 Tromsø, Norway 2 Institute of Biology, Komi Science Centre, Ural Division, Russian Academy of Sciences, 28 Kommunisticheskaya str., 167982, Syktyvkar, Republic of Komi, Russia Corresponding author : * [email protected] ABSTRACT Maral root (Leuzea charthamoides DC), roseroot (Rhodiola rosea L.), and crowned saw-wort (Serratula coronata L.) were grown in a phytotron under controlled conditions at 9, 15, 21°C day/9°C night and 21°C. All these treatments had 24 hours of light (long day-LD). In addition there was one treatment at 21°C with only 12 hours of light (short day-SD). Plants were harvested after four months and plant growth was recorded. Leaves of S. coronata and the underground part of L. carthamoides and R. rosea were dried and analyzed for adaptogenic compounds. The number of shoots and dry weight of caudex with roots of R. rosea increased by raising the temperature from 9 to 15°C. Differentiated day and night temperature with an average temperature of 15°C further increased the growth. The lowest number of shoots and the lowest dry weight of roots were produced at the highest temperature (21°C). The concentration of tyrosol and cinnamic alcohol in dried R. -

Study on Callus Production and Plant Regeneration of Leuzea
Vol. 8(5), pp. 260-268, 3 February, 2014 DOI: 10.5897/JMPR11.610 ISSN 1996-0875 ©2014 Academic Journals Journal of Medicinal Plants Research http://www.academicjournals.org/JMPR Full Length Research Paper Study on callus induction and plant regeneration of Leuzea carthamoides via tissue culture system Akhtar Zand, Alireza Babaei*, Reza Omidbaigi and Elham Daneshfar Department of Horticulture, College of Agriculture, Tarbiat Modares University, Tehran, Islamic Republic of Iran. Accepted 23 May, 2011 Leuzea (Rhaponticum carthamoides) is a valuable medicinal plant from Asteraceae. Micropropagation could be a good alternative for the mass propagation of Leuzea carthamoides. To investigate the callogenesis of leaf explants, 12 different hormonal combinations including different concentrations of 16-benzylaminopurine (BA) and 2,4-dichlorophenoxyacetic acid (2, 4-D) were studied in two separable experiments. In both experiments, the explants were transferred to the Ms medium supplemented with 0.5 mg L-1 indole acetic acid (IAA) and 0.5 mg L-1 BA for 7 and 50 days after culture for regeneration, respectively. Then, after one month the percentages of callogenesis and the amount of produced callus were measured. In other experiment to investigated regeneration of root explants, 9 different hormonal combinations were studied including different concentrations of BA and IAA. The number of leaf per explants, length of greatest leaf per explant and regeneration percentage were measured one month after culture. The maximum callus production was obtained using 1 mg L-1 2, 4-D and 1.5 mg L-1 BA and 0.25 mg L-1 2, 4-D and 1.5 mg L-1 BA in first experiment and second experiment, respectively. -

Acute and Chronic Effects of Rhaponticum Carthamoides and Rhodiola Rosea Extracts Supplementation Coupled to Resistance Exercise
Roumanille et al. Journal of the International Society of Sports Nutrition (2020) 17:58 https://doi.org/10.1186/s12970-020-00390-5 RESEARCH ARTICLE Open Access Acute and chronic effects of Rhaponticum carthamoides and Rhodiola rosea extracts supplementation coupled to resistance exercise on muscle protein synthesis and mechanical power in rats Rémi Roumanille1* , Barbara Vernus1, Thomas Brioche1, Vincent Descossy1, Christophe Tran Van Ba1, Sarah Campredon1, Antony G. Philippe1,2, Pierre Delobel1, Christelle Bertrand-Gaday1, Angèle Chopard1, Anne Bonnieu1, Guillaume Py1 and Pascale Fança-Berthon3 Abstract Background: Owing to its strength-building and adaptogenic properties, Rhaponticum carthamoides (Rha) has been commonly used by elite Soviet and Russian athletes. Rhodiola rosea (Rho) is known to reduce physical and mental fatigue and improve endurance performance. However, the association of these two nutritional supplements with resistance exercise performance has never been tested. Resistance exercise is still the best way to stimulate protein synthesis and induce chronic muscle adaptations. The aim of this study was to investigate the acute and chronic effects of resistance exercise coupled with Rha and Rho supplementation on protein synthesis, muscle phenotype, and physical performance. Methods: For the acute study, fifty-six rats were assigned to either a trained control group or one of the groups treated with specific doses of Rha and/or Rho. Each rats performed a single bout of climbing resistance exercise. The supplements were administered immediately after exercise by oral gavage. Protein synthesis was measured via puromycin incorporation. For the chronic study, forty rats were assigned to either the control group or one of the groups treated with doses adjusted from the acute study results. -

Index Seminum Et Sporarum Perm, 2013
INDEX SEMINUM ET SPORARUM QUAE HORTUS BOTANICUS UNIVERSITATIS BIARMIENSIS PRO MUTUA COMMUTATIONE OFFERT СПИСОК СЕМЯН И СПОР , предлагаемых для обмена Ботаническим садом имени проф . А.Г. Генкеля Пермского государственного национального исследовательского университета Пермь , Россия Biarmiae 2013 Federal State Budgetary Educational Institution of Higher Professional Education «Perm State University», Botanic Garden ______________________________________________________________________________________ Дорогие друзья ботанических садов , Дорогие коллеги ! Ботанический сад Пермского государственного национального исследовательского университета был создан в 1922 г. по инициативе и под руководством проф . А.Г. Генкеля . Здесь работали известные ученые – ботаники Д.А. Сабинин , В.И. Баранов , Е.А. Павский , внесшие своими исследованиями большой вклад в развитие биологических наук на Урале . В настоящее время Ботанический сад имени профессора А.Г. Генкеля входит в состав регионального Совета ботанических садов Урала и Поволжья , имеет статус научного учреждения и особо охраняемой природной территории . Основными научными направлениями работы являются : интродукция и акклиматизация растений , выведение и отбор новых форм и сортов , наиболее стойких и продуктивных в местных условиях . Ботанический сад расположен на двух участках общей площадью 2,7 га . Коллекции включают около 4000 видов , форм и сортов древесных , кустарниковых и травянистых растений , произрастающих в открытом и закрытом грунте . Из оранжерейных растений полнее всего представлены -

Ecdysteroid Effects on Algae
COMPILATION OF THE LITERATURE REPORTS FOR THE EFFECTS OF ECDYSTEROIDS ON ALGAE, VASCULAR PLANTS, MICROBES, INSECTS AND MAMMALS, THEIR BIOTECHNOLOGICAL APPLICATIONS AND THEIR BIOLOGICAL ACTIVITIES Compiled by Laurie Dinan and René Lafont, Sorbonne Universités – UPMC Université Paris 06, IBPS-BIOSIPE, Case Courrier 29, 7 Quai St. Bernard, F-75252 Paris Cedex 05, France. Version 3: Date of last update: 21/04/17 Important notice: This database has been designed as a tool to help the scientific community in research on ecdysteroids. The authors wish it to be an evolving system and would encourage other researchers to submit new data, additional publications, proposals for modifications or comments to the authors for inclusion. All new material will be referenced to its contributor. Reproduction of the material in this database in its entirety is not permitted. Reproduction of parts of the database is only permitted under the following conditions: reproduction is for personal use, for teaching and research, but not for distribution to others reproduction is not for commercial use the origin of the material is indicated in the reproduction we should be notified in advance to allow us to document that the reproduction is being made Where data are reproduced in published texts, they should be acknowledged by the reference: Lafont R., Harmatha J., Marion-Poll F., Dinan L., Wilson I.D.: The Ecdysone Handbook, 3rd edition, on-line, http://ecdybase.org Illustrations may not under any circumstances be used in published texts, commercial or otherwise, without previous written permission of the author(s). Please notify Laurie Dinan ([email protected]) of any errors or additional literature sources. -

Pharmaceutical Botany
PHARMACEUTICAL BOTANY In two parts Part 1 Student of 1st year _______ group ____________________________________________________________ (Full name) Minsk BSMU 2018 МИНИСТЕРСТВО ЗДРАВООХРАНЕНИЯ РЕСПУБЛИКИ БЕЛАРУСЬ БЕЛОРУССКИЙ ГОСУДАРСТВЕННЫЙ МЕДИЦИНСКИЙ УНИВЕРСИТЕТ КАФЕДРА ОРГАНИЗАЦИИ ФАРМАЦИИ ФАРМАЦЕВТИЧЕСКАЯ БОТАНИКА PHARMACEUTICAL BOTANY Практикум для специальности «Фармация» В двух частях Часть 1 Минск БГМУ 2018 УДК 615.1:58(076.5)(075.8)-054.6 ББК 52.82:28.5я73 Ф24 Рекомендовано Научно-методическим советом университета в качестве практикума 21.02.2018 г., протокол № 6 А в т о р ы: О. А. Кузнецова, Н. С. Гурина, М. В. Волочник, Н. М. Борабанова Р е ц е н з е н т ы: канд. мед. наук, доц. Белорусского государственного медицинского университета Л. М. Сычик; канд. мед. наук, доц. Витебского государственного ордена Дружбы народов медицинского университета Л. А. Любаковская Фармацевтическая ботаника = Pharmaceutical Botany : практикум для специальности «Фармация». В 2 ч. Ч. 1 / О. А. Кузнецова Ф24 [и др.]. – Минск : БГМУ, 2018. – 66 с. ISBN 978-985-567-970-8. Включены контрольные вопросы, основные термины и понятия; закрытые и открытые тесты для самоконтроля; рисунки, таблицы и задания по ботанике и си- стематике растений. Предназначен для студентов 1-го курса медицинского факультета иностранных учащихся, обучающихся на английском языке по специальности «Фармация». УДК 615.1:58(076.5)(075.8)-054.6 ББК 52.82:28.5я73 ISBN 978-985-567-970-8 (Ч. 1) © УО «Белорусский государственный ISBN 978-985-567-971-5 медицинский университет», 2018 Учебное издание Кузнецова Ольга Анатольевна Гурина Наталия Сергеевна Волочник Мария Валерьевна Борабанова Надежда Михайловна ФАРМАЦЕВТИЧЕСКАЯ БОТАНИКА PHARMACEUTICAL BOTANY Практикум для специальности «Фармация» На английском языке В двух частях Часть 1 Ответственная за выпуск О. -

Research Article Establishment of Hairy Root Cultures of Rhaponticum Carthamoides (Willd.) Iljin for the Production of Biomass and Caffeic Acid Derivatives
Hindawi Publishing Corporation BioMed Research International Volume 2015, Article ID 181098, 11 pages http://dx.doi.org/10.1155/2015/181098 Research Article Establishment of Hairy Root Cultures of Rhaponticum carthamoides (Willd.) Iljin for the Production of Biomass and Caffeic Acid Derivatives Ewa SkaBa,1 Agnieszka Kicel,2 Monika A. Olszewska,2 Anna K. Kiss,3 and Halina WysokiNska1 1 Department of Biology and Pharmaceutical Botany, Medical University of Łod´ z,´ Muszynskiego´ 1, 90-151 Łod´ z,´ Poland 2Department of Pharmacognosy, Medical University of Łod´ z,´ Muszynskiego´ 1, 90-151 Łod´ z,´ Poland 3Department of Pharmacognosy and Molecular Basis of Phytotherapy, Medical University of Warsaw, Banacha1,02-097Warsaw,Poland Correspondence should be addressed to Ewa Skała; [email protected] Received 27 December 2014; Accepted 5 February 2015 Academic Editor: Alberto Reis Copyright © 2015 Ewa Skała et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. The aim of the study was to obtain transformed roots of Rhaponticum carthamoides and evaluate their phytochemical profile. Hairy roots were induced from leaf explants by the transformation of Agrobacterium rhizogenes strains A4 and ATCC 15834. The best response (43%) was achieved by infection with A4 strain. The effects of different liquid media (WPM, B5, SH) with fulland half-strength concentrations of macro- and micronutrients on biomass accumulation of the best grown hairy root line (RC3) at −1 two different lighting conditions (light or dark) were investigated. The highest biomass (93 gL of the fresh weight after 35 days) 3 was obtained in WPM medium under periodic light. -

Asteraceae, Cardueae)
UNIVERSITAT DE BARCELONA. FACULTAT DE FARMÀCIA Departament de Productes Naturals, Biologia Vegetal i Edafologia, Secció Botànica Programa de Doctorat: Biodiversitat 2009-2012 INSTITUT BOTÀNIC DE BARCELONA (IBB-CSIC-ICUB) Estudio biosistemático y evolutivo del género Echinops L. (Asteraceae, Cardueae) Memòria presentada per Ismael Sánchez Jiménez per a optar al títol de Doctor per la Universitat de Barcelona Amb el vist-i-plau de les directores de tesi: Dra. Teresa Garnatje Roca Dra. Oriane Hidalgo i del tutor de tesi: Dr. Joan Vallès Xirau Ismael Sánchez Jiménez Barcelona, 2012 ¿Por qué subir al Everest? Porque está ahí... George Mallory ÍNDICE Agradecimientos 7 Introducción 11 1. Marco taxonómico 11 1.1. La familia Asteraceae Martinov 11 1.2. La tribu Cardueae Cass. 15 1.3. La subtribu Echinopsinae (Cass.) Dumort. y el género Echinops L. 17 2. Antecedentes 23 2.1. Recuentos cromosómicos y cantidad de ADN 23 2.2. Palinología 25 2.3. Sistemática molecular 29 Objetivos de la Tesis Doctoral 33 Informe de las directoras de la Tesis 35 Síntesis y discusión de los resultados 39 Conclusiones 51 Referencias 53 Compendio de publicaciones 63 Chromosome Numbers in Three Asteraceae Tribes from Inner Mongolia (China), with Genome Size Data for Cardueae 65 Molecular systematics of Echinops L. (Asteraceae, Cynareae): A phylogeny based on ITS and trnL-trnF sequences with emphasis on sectional delimitation 83 Genome size and chromosome number in Echinops (Asteraceae, Cardueae) in the Aegean and Balkan regions: technical aspects of nuclear DNA amount assessment and genome evolution in a phylogenetic frame 99 Echinops spinosissimus Turra subsp. neumayeri (Vis.) Kožuharov (Asteraceae, Cardueae): a new record for the flora of Greece 117 Pollen study in the genus Echinops L. -

Anatomy of Subterranean Organs of Medicinally Used Cardueae and Related Species and Its Value for Discrimination
Sci Pharm www.scipharm.at Research article Open Access Anatomy of Subterranean Organs of Medicinally Used Cardueae and Related Species and its Value for Discrimination Elisabeth FRITZ *, Johannes SAUKEL Department of Pharmacognosy, University of Vienna, Althanstrasse 14, 1090, Vienna, Austria * Corresponding author. E-mail: [email protected] (E. Fritz) Sci Pharm. 2011; 79: 157–174 doi:10.3797/scipharm.1010-05 Published: December 2nd 2010 Received: October 20th 2010 Accepted: December 2nd 2010 This article is available from: http://dx.doi.org/10.3797/scipharm.1010-05 © Fritz and Saukel et al.; licensee Österreichische Apotheker-Verlagsgesellschaft m. b. H., Vienna, Austria. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/3.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Numerous species of the Asteraceae, the composites, are famous for their use in both traditional and conventional medicine. Reliable anatomical descriptions of these plants and of possible adulterations provide a basis for fast identification and cheap purity controls of respective medicinal drugs by means of light microscopy. Nevertheless, detailed comparative studies on root and rhizome anatomy of valuable as well as related inconsiderable composite plants are largely missing yet. The presented study aims to narrow this gap by performing anatomical analyses of roots and rhizomes