(ALDH7A1) Attenuates Reactive Aldehyde and Oxidative Stress Induced Cytotoxicity
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

(ALDH1A3) for the Maintenance of Non-Small Cell Lung Cancer Stem Cells Is Associated with the STAT3 Pathway
Author Manuscript Published OnlineFirst on June 6, 2014; DOI: 10.1158/1078-0432.CCR-13-3292 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Essential role of aldehyde dehydrogenase 1A3 (ALDH1A3) for the maintenance of non-small cell lung cancer stem cells is associated with the STAT3 pathway Chunli Shao1,2, James P. Sullivan3, Luc Girard1,2, Alexander Augustyn1,2, Paul Yenerall1,2, Jaime Rodriguez4, Hui Liu4, Carmen Behrens4, Jerry W. Shay5, Ignacio I. Wistuba4, John D. Minna 1,2,6,7 1Hamon Center for Therapeutic Oncology Research, 2Simmons Comprehensive Cancer Center, 3Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts, 02114, 4Department of Translational Molecular Pathology, University of Texas M.D. Anderson Cancer Center, Houston, Texas, 77054, 5Department of Cell Biology, 6Department of Pharmacology, 7Internal Medicine, University of Texas Southwestern Medical Center, Dallas, Texas, 75390, USA. Running Title: ALDH1A3 in non-small cell lung cancer stem cells Keywords: Lung cancer, cancer stem cells, ALDH1A3, STAT3, Stattic Financial Support This project was supported by CPRIT, NCI SPORE P50CA70907, UTSW Cancer Center Support Grant 5P30-CA142543, and the Gillson-Longenbaugh Foundation. Address Correspondence: John D. Minna, M.D. 6000 Harry Hines Blvd Dallas, TX 75390-8593 Hamon Center for Therapeutic Oncology Research UT Southwestern Medical Center Phone: 214-648-4900; Fax: 214-648-4940 [email protected] Disclosure of Potential Conflict of Interest The authors indicate no potential conflicts of interest. Word count: 4583 Total number of figures and tables: 6 figures Downloaded from clincancerres.aacrjournals.org on September 28, 2021. © 2014 American Association for Cancer Research. -

Overall Survival of Pancreatic Ductal Adenocarcinoma Is Doubled by Aldh7a1 Deletion in the KPC Mouse
Overall survival of pancreatic ductal adenocarcinoma is doubled by Aldh7a1 deletion in the KPC mouse Jae-Seon Lee1,2*, Ho Lee3*, Sang Myung Woo4, Hyonchol Jang1, Yoon Jeon1, Hee Yeon Kim1, Jaewhan Song2, Woo Jin Lee4, Eun Kyung Hong5, Sang-Jae Park4, Sung- Sik Han4§§ and Soo-Youl Kim1§ 1Division of Cancer Biology, Research Institute, National Cancer Center, Goyang, Republic of Korea. 2Department of Biochemistry, College of Life Science and Biotechnology, Yonsei University, Seoul, Republic of Korea. 3Graduate School of Cancer Science and Policy, 4Department of Surgery, Center for Liver and Pancreatobiliary Cancer and 5Department of Pathology, National Cancer Center, Goyang, Republic of Korea. Correspondence §Corresponding author: [email protected] (S.-Y.K.) §§Co-corresponding author: [email protected] (S.-S.H.) *These authors contributed equally to this work 1 Abstract Rationale: The activity of aldehyde dehydrogenase 7A1 (ALDH7A1), an enzyme that catalyzes the lipid peroxidation of fatty aldehydes was found to be upregulated in pancreatic ductal adenocarcinoma (PDAC). ALDH7A1 knockdown significantly reduced tumor formation in PDAC. We raised a question how ALDH7A1 contributes to cancer progression. Methods: To answer the question, the role of ALDH7A1 in energy metabolism was investigated by knocking down and knockdown gene in mouse model, because the role of ALDH7A1 has been reported as a catabolic enzyme catalyzing fatty aldehyde from lipid peroxidation to fatty acid. Oxygen consumption rate (OCR), ATP production, mitochondrial membrane potential, proliferation assay and immunoblotting were performed. In in vivo study, two human PDAC cell lines were used for pre-clinical xenograft model as well as spontaneous PDAC model of KPC mice was also employed for anti-cancer therapeutic effect. -

Insights Into Aldehyde Dehydrogenase Enzymes: a Structural Perspective
REVIEW published: 14 May 2021 doi: 10.3389/fmolb.2021.659550 Insights into Aldehyde Dehydrogenase Enzymes: A Structural Perspective Kim Shortall, Ahmed Djeghader, Edmond Magner and Tewfik Soulimane* Department of Chemical Sciences, Bernal Institute, University of Limerick, Limerick, Ireland Aldehyde dehydrogenases engage in many cellular functions, however their dysfunction resulting in accumulation of their substrates can be cytotoxic. ALDHs are responsible for the NAD(P)-dependent oxidation of aldehydes to carboxylic acids, participating in detoxification, biosynthesis, antioxidant and regulatory functions. Severe diseases, including alcohol intolerance, cancer, cardiovascular and neurological diseases, were linked to dysfunctional ALDH enzymes, relating back to key enzyme structure. An in-depth understanding of the ALDH structure-function relationship and mechanism of action is key to the understanding of associated diseases. Principal structural features 1) cofactor binding domain, 2) active site and 3) oligomerization mechanism proved critical in maintaining ALDH normal activity. Emerging research based on the combination of structural, functional and biophysical studies of bacterial and eukaryotic ALDHs contributed to the appreciation of diversity within the superfamily. Herewith, we Edited by: discuss these studies and provide our interpretation for a global understanding of Ashley M Buckle, ALDH structure and its purpose–including correct function and role in disease. Our Monash University, Australia analysis provides a synopsis -

A Novel ALDH1A1 Inhibitor Targets Cells with Stem Cell Characteristics in Ovarian Cancer
cancers Article A Novel ALDH1A1 Inhibitor Targets Cells with Stem Cell Characteristics in Ovarian Cancer Nkechiyere G. Nwani 1, Salvatore Condello 2 , Yinu Wang 1, Wendy M. Swetzig 1, Emma Barber 1, Thomas Hurley 3,4 and Daniela Matei 1,5,6,* 1 Department of Obstetrics and Gynecology, Northwestern University, Chicago, IL 60611, USA; [email protected] (N.G.N.); [email protected] (Y.W.); [email protected] (W.M.S.); [email protected] (E.B.) 2 Department of Obstetrics and Gynecology, Indiana University, Indianapolis, IN 46202, USA; [email protected] 3 Department of Biochemistry and molecular Biology, Indiana University, Indianapolis, IN 46202, USA; [email protected] 4 Melvin and Bren Simon Cancer Center, Indianapolis, IN 46202, USA 5 Robert H Lurie Comprehensive Cancer Center, Chicago, IL 60611, USA 6 Jesse Brown VA Medical Center, Chicago, IL 60612, USA * Correspondence: [email protected] Received: 8 February 2019; Accepted: 3 April 2019; Published: 8 April 2019 Abstract: A small of population of slow cycling and chemo-resistant cells referred to as cancer stem cells (CSC) have been implicated in cancer recurrence. There is emerging interest in developing targeted therapeutics to eradicate CSCs. Aldehyde-dehydrogenase (ALDH) activity was shown to be a functional marker of CSCs in ovarian cancer (OC). ALDH activity is increased in cells grown as spheres versus monolayer cultures under differentiating conditions and in OC cells after treatment with platinum. Here, we describe the activity of CM37, a newly identified small molecule with inhibitory activity against ALDH1A1, in OC models enriched in CSCs. Treatment with CM37 reduced OC cells’ proliferation as spheroids under low attachment growth conditions and the expression of stemness-associated markers (OCT4 and SOX2) in ALDH+ cells fluorescence-activated cell sorting (FACS)-sorted from cell lines and malignant OC ascites. -

Aldehyde Dehydrogenase, Liver Disease and Cancer Wenjun Wang1, Chunguang Wang2, Hongxin Xu1, Yanhang Gao1
Int. J. Biol. Sci. 2020, Vol. 16 921 Ivyspring International Publisher International Journal of Biological Sciences 2020; 16(6): 921-934. doi: 10.7150/ijbs.42300 Review Aldehyde Dehydrogenase, Liver Disease and Cancer Wenjun Wang1, Chunguang Wang2, Hongxin Xu1, Yanhang Gao1 1. Department of Hepatology, The First Hospital of Jilin University, Jilin University, Changchun, Jilin, 130021, China. 2. Department of Thoracic & Cardiovascular Surgery, Second Clinical College, Jilin University, Changchun, 130041, China. Corresponding author: Yanhang Gao, MD., PhD., Department of Hepatology, The First Hospital of Jilin University, Jilin University, Changchun, Jilin, 130021, China. Email: [email protected]. Tel: +86 15804303019; +86 431 81875121; +86 431 81875106; Fax number: 0431-81875106. © The author(s). This is an open access article distributed under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0/). See http://ivyspring.com/terms for full terms and conditions. Received: 2019.11.20; Accepted: 2020.01.03; Published: 2020.01.22 Abstract Acetaldehyde dehydrogenase 2 (ALDH2) is the key enzyme responsible for metabolism of the alcohol metabolite acetaldehyde in the liver. In addition to conversion of the acetaldehyde molecule, ALDH is also involved in other cellular functions. Recently, many studies have investigated the involvement of ALDH expression in viral hepatitis, alcoholic liver disease (ALD), non-alcoholic fatty liver disease (NAFLD), liver fibrosis, and liver cancer. Notably, ALDH2 expression has been linked with liver cancer risk, as well as pathogenesis and prognosis, and has emerged as a promising therapeutic target. Of note, approximately 8% of the world’s population, and approximately 30-40% of the population in East Asia carry an inactive ALDH2 gene. -

ALDH1A2 Is a Candidate Tumor Suppressor Gene in Ovarian Cancer
Supplementary Materials ALDH1A2 is a Candidate Tumor Suppressor Gene in Ovarian Cancer Jung-A Choi, Hyunja Kwon, Hanbyoul Cho*, Joon-Yong Chung, Stephen M. Hewitt and Jae-Hoon Kim Figure S1. Transfection efficiency after pCMV-tagB-Flag-ALDH1A2 transfection in RMG-I, SKOV3, and OVCA433 cells. Cells were grown on coverslips and transfected with pCMV-tagB-Flag- ALDH1A2 using Lipofectamine® 2000. Cells were then immunostained with primary antibodies for Flag and visualized by confocal microscopy (A). Transfection efficiency (%) was evaluated by counting cells expressing Flag. Figure S2. Effect of ALDH1A2 overexpression on cell cycle distribution and cell death in ovarian cancer. (A-B) Cells were transfected with pCMV-tagB-Flag-ALDH1A2 using Lipofectamine® 2000. After 24 h, the cells were harvested, fixed in ice-cold 70% ethanol, and stained with propidium iodide. Cell-cycle distribution (A) and cell death (B) were analyzed by fluorescence-activated cell sorting (FACS). Apoptotic cells were quantified for DNA content after propidium iodide staining; the sub- G 1 fraction (%) represents the proportion of apoptotic cells. Statistical significance was assessed using an unpaired t test. **p < 0.05, ***p < 0.005 . 1 Figure S3. Annexin V/PI staining in RMG-I, SKOV3, and OVCA433 cells overexpressing ALDH1A2. (A-B) Cells were transfected with pCMV-tagB-Flag-ALDH1A2 using Lipofectamine® 2000. After the indicated time, cells were immediately stained with Annexin V-FITC and PI, subjected to flow cytometry analyses (A). Quantitative results of cell death were determined using Annexin V/PI (B). 2 Figure S4. Methylation status of ALDH1A2 genes in public datasets, Methylation and Expression Database of Normal and Tumor Tissues (MENT; http://mgrc.kribb.re.kr:8080/MENT/). -

Cells in Diabetic Mice
ARTICLE Received 12 Jan 2016 | Accepted 18 Jul 2016 | Published 30 Aug 2016 DOI: 10.1038/ncomms12631 OPEN Aldehyde dehydrogenase 1a3 defines a subset of failing pancreatic b cells in diabetic mice Ja Young Kim-Muller1,*, Jason Fan1,2,*, Young Jung R. Kim2, Seung-Ah Lee1, Emi Ishida1, William S. Blaner1 & Domenico Accili1 Insulin-producing b cells become dedifferentiated during diabetes progression. An impaired ability to select substrates for oxidative phosphorylation, or metabolic inflexibility, initiates progression from b-cell dysfunction to b-cell dedifferentiation. The identification of pathways involved in dedifferentiation may provide clues to its reversal. Here we isolate and functionally characterize failing b cells from various experimental models of diabetes and report a striking enrichment in the expression of aldehyde dehydrogenase 1 isoform A3 (ALDH þ )asb cells become dedifferentiated. Flow-sorted ALDH þ islet cells demonstrate impaired glucose- induced insulin secretion, are depleted of Foxo1 and MafA, and include a Neurogenin3- positive subset. RNA sequencing analysis demonstrates that ALDH þ cells are characterized by: (i) impaired oxidative phosphorylation and mitochondrial complex I, IV and V; (ii) activated RICTOR; and (iii) progenitor cell markers. We propose that impaired mitochondrial function marks the progression from metabolic inflexibility to dedifferentiation in the natural history of b-cell failure. 1 Naomi Berrie Diabetes Center and Department of Medicine, Columbia University, New York, New York 10032, USA. 2 Department of Genetics and Integrated Program in Cellular, Molecular and Biomedical Studies, Columbia University, New York, New York 10032, USA. * These authors contributed equally to this work. Correspondence and requests for materials should be addressed to D.A. -
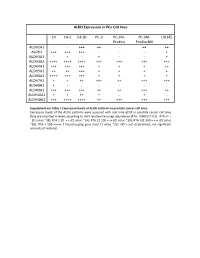
ALDH Expression in Pca Cell Lines C4 C4-2 C4-2B PC-3 PC-3M- Pro4luc PC-3M- Pro4lucbiii DU145 ALDH1A1
ALDH Expression in PCa Cell lines C4 C4-2 C4-2B PC-3 PC-3M- PC-3M- DU145 Pro4luc Pro4lucBIII ALDH1A1 - - +++ ++ - ++ ++ ALDH2 +++ +++ +++ - - - + ALDH3A1 - + - + - - + ALDH3A2 ++++ ++++ ++++ +++ +++ +++ +++ ALDH4A1 +++ +++ +++ + + + ++ ALDH5A1 ++ ++ +++ + + + + ALDH6A1 ++++ +++ +++ + + + + ALDH7A1 + + ++ +++ ++ +++ +++ ALDH8A1 + - - - - - - ALDH9A1 +++ +++ +++ ++ ++ +++ ++ ALDH16A1 + + ++ + - + - ALDH18A1 +++ ++++ ++++ ++ +++ +++ +++ Supplementary Table I. Expression levels of ALDH isoforms in prostate cancer cell lines. Expression levels of the ALDH isoforms were assessed with real time qPCR in prostate cancer cell lines Data are classified in levels according to their relative transcript abundance (RTA: 10000/2^ΔCt). RTA <1 = - (Ct value ~38); RTA 1-20 = + (Ct value ~34); RTA 21-100 = ++ (Ct value ~30); RTA 101-500 = +++ (Ct value ~26); RTA > 500 =++++ ( housekeeping gene level Ct value ~22). ND = not determined, not-significant amounts of material. ALDH Expression in Prostate Cancer PCa primary tumor PCa primary cultures #44 #99 #62#69 123 583 697 233 549 069B 567B ALDH1A1 ++ + ND ND ++++ ND ND ND ND ND ND ALDH2 + ++ ND ND ++++ ND ND ND ND ND ND ALDH3A1 ++++ ++++ +++ ++ ++ +++ ++++ ++++ +++ ++ + ALDH3A2 +++ +++ +++ ++++ + ++++ ++++ ++++ ++++ ++++ ++++ ALDH4A1 ++ +++ ++ ++ +++ ND ND ND ND ND ND ALDH5A1 ++ +++ ++ ++ - ND ND ND ND ND ND ALDH6A1 ++ + ND ND ++++ ND ND ND ND ND ND ALDH7A1 ++++ ++++ +++ +++ ++++ ++++ ++++ ++++ ++++ ++++ ++++ ALDH8A1 + + ND ND + ND ND ND ND ND ND ALDH9A1 ++ ++ ND ND ++++ ND ND ND ND ND ND ALDH16A1 +++ ++ ND ND ++++ ND ND ND ND ND ND ALDH18A1 - - +++ +++ - ++++ ++++ ++++ ++++ +++++++ Supplementary Table II. Expression levels of ALDH isoforms in primary prostate cancer tissue samples and primary cultures. Expression levels of the ALDH isoforms were assessed with real time qPCR in primary prostate cancer tissue samples (left panel) and primary cultures (right panel). Data are classified in levels according to their relative transcript abundance. -

Genotypic and Phenotypic Spectrum of Pyridoxine-Dependent Epilepsy (ALDH7A1 Deficiency) Downloaded from Philippa B
doi:10.1093/brain/awq143 Brain 2010: 133; 2148–2159 | 2148 BRAIN A JOURNAL OF NEUROLOGY Genotypic and phenotypic spectrum of pyridoxine-dependent epilepsy (ALDH7A1 deficiency) Downloaded from Philippa B. Mills,1 Emma J. Footitt,1 Kevin A. Mills,1 Karin Tuschl,1 Sarah Aylett,1 Sophia Varadkar,1 Cheryl Hemingway,1 Neil Marlow,2 Janet Rennie,2 Peter Baxter,3 Olivier Dulac,4 Rima Nabbout,4 William J. Craigen,5 Bernhard Schmitt,6 Franc¸ois Feillet,7 Ernst Christensen,8 Pascale De Lonlay,9 Mike G. Pike,10 M. Imelda Hughes,11 Eduard A. Struys,12 12 13 1 Cornelis Jakobs, Sameer M. Zuberi and Peter T. Clayton http://brain.oxfordjournals.org 1 Institute of Child Health, University College London with Great Ormond Street Hospital for Children, National Health Service Trust, London, UK 2 Division of Neonatal Medicine, EGA UCL Institute for Women’s Health, University College Hospital, London WC1N 1EH, UK 3 Department of Paediatric Neurology, Sheffield Children’s National Health Service Foundation Trust, Sheffield S10 5DD, UK 4 Department of Neuropaediatrics APHP, Centre de Re´ fe´ rence E´ pilepsies Rares, Necker-Enfants Malades Hospital, 75743 Paris, Cedex 15, France 5 Department of Molecular and Human Genetics, Baylor College of Medicine, One Baylor Plaza MS BCM225 Houston, TX, 77030, USA 6 Department of Neurology, University Children’s Hospital, Steinwiesstrasse 75, CH-8032 Zurich, Switzerland 7 Centre de Re´ fe´ rence des Maladies He´ re´ ditaires du Me´ tabolisme et INSERM U954, CHU Brabois Enfants, et Faculte´ de Me´ decine de Nancy, Alle´ e du -

Novel Protein Pathways in Development and Progression of Pulmonary Sarcoidosis Maneesh Bhargava1*, K
www.nature.com/scientificreports OPEN Novel protein pathways in development and progression of pulmonary sarcoidosis Maneesh Bhargava1*, K. J. Viken1, B. Barkes2, T. J. Grifn3, M. Gillespie2, P. D. Jagtap3, R. Sajulga3, E. J. Peterson4, H. E. Dincer1, L. Li2, C. I. Restrepo2, B. P. O’Connor5, T. E. Fingerlin5, D. M. Perlman1 & L. A. Maier2 Pulmonary involvement occurs in up to 95% of sarcoidosis cases. In this pilot study, we examine lung compartment-specifc protein expression to identify pathways linked to development and progression of pulmonary sarcoidosis. We characterized bronchoalveolar lavage (BAL) cells and fuid (BALF) proteins in recently diagnosed sarcoidosis cases. We identifed 4,306 proteins in BAL cells, of which 272 proteins were diferentially expressed in sarcoidosis compared to controls. These proteins map to novel pathways such as integrin-linked kinase and IL-8 signaling and previously implicated pathways in sarcoidosis, including phagosome maturation, clathrin-mediated endocytic signaling and redox balance. In the BALF, the diferentially expressed proteins map to several pathways identifed in the BAL cells. The diferentially expressed BALF proteins also map to aryl hydrocarbon signaling, communication between innate and adaptive immune response, integrin, PTEN and phospholipase C signaling, serotonin and tryptophan metabolism, autophagy, and B cell receptor signaling. Additional pathways that were diferent between progressive and non-progressive sarcoidosis in the BALF included CD28 signaling and PFKFB4 signaling. Our studies demonstrate the power of contemporary proteomics to reveal novel mechanisms operational in sarcoidosis. Application of our workfows in well-phenotyped large cohorts maybe benefcial to identify biomarkers for diagnosis and prognosis and therapeutically tenable molecular mechanisms. -

Supplemental Information
Supplemental Information Aldehyde dehydrogenases and prostate cancer: shedding light on isoform distribution to reveal druggable target. Luca Quattrini, Maria Sadiq, Giovanni Petrarolo, Norman J. Maitland, Fiona M. Frame, Klaus Pors, Concettina la Motta. Table of Contents Table S1. Taqman Assay details. Figure S1. Synthetic procedure used to derive (substituted)imidazo[1,2-a]pyridine derivatives 3a-d. Figure S2. qPCR analysis of ALDH expression in basal epithelial cells after subpopulation selection. Figure S3. Western Blot analysis of ALDH1A1 and ALDH1A3 expression in prostate cell lines and primary prostate epithelial cells. Figure S4: ImageJ analysis of fluorescence intensity profile of ALDH1A1 and ALDH1A3 staining. Figure S5. ALDH gene expression analysis of primary prostatic epithelial cells after atRA treatment. S1 Table S1: Taqman Assay details S2 Figure S1. Synthetic procedure used to derive (substituted)imidazo[1,2-a]pyridine derivatives 3a-d S3 Figure S2. qPCR analysis of ALDH expression in basal epithelial cells after subpopulation selection. Gene expression of (A) ALDH1A1, (B) ALDH1A2 and (C) ALDH1A3, (D) ALDH1B1, (E) ALDH2, (F) ALDH3A1 and (G) ALDH7A1 using 2-dCT. RPLP0 was used as the control gene. SC- stem cell, TA- transit amplifying cell, and CB- committed basal cells. For ALDH1A1, 1A2, -2, and -3A1 statistical significance was assessed using the Mann Whitney U test for comparing unpaired groups for TA compared with CB, herein SC (n=1), TA (n=3) and CB (n=4). Due to only one sample for SC, S4 statistical test for this population was unobtainable. For ALDH1A3, -1B1 and -7A1 statistical significance was assessed using the one-way ANOVA for multiple comparisons of unpaired groups, TA (n=8) and CB (n=8) compared to SC (n=3), and no statistical significance was found. -

Aldehyde Dehydrogenases and Prostate Cancer: Shedding Light on Isoform Distribution to Reveal Druggable Target
biomedicines Article Aldehyde Dehydrogenases and Prostate Cancer: Shedding Light on Isoform Distribution to Reveal Druggable Target 1, 2, 1 3 Luca Quattrini y, Maria Sadiq y, Giovanni Petrarolo , Norman J. Maitland , Fiona M. Frame 3, Klaus Pors 2,* and Concettina La Motta 1,4,* 1 Department of Pharmacy, University of Pisa, Via Bonanno 6, 56126 Pisa, Italy; [email protected] (L.Q.); [email protected] (G.P.) 2 Institute of Cancer Therapeutics, School of Pharmacy and Medical Sciences, Faculty of Life Sciences, University of Bradford, West Yorkshire BD7 1DP, UK; [email protected] 3 Cancer Research Unit, Department of Biology, University of York, Heslington, North Yorkshire YO10 5DD, UK; [email protected] (N.J.M.); fi[email protected] (F.M.F.) 4 CISUP, Centre for Instrumentation Sharing, University of Pisa, Lungarno Pacinotti 43, 56128 Pisa, Italy * Correspondence: [email protected] (K.P.); [email protected] (C.L.M.) Contribution to the work: L.Q. and M.S. contributed equally. y Received: 22 October 2020; Accepted: 1 December 2020; Published: 4 December 2020 Abstract: Prostate cancer represents the most common malignancy diagnosed in men, and is the second-leading cause of cancer death in this population. In spite of dedicated efforts, the current therapies are rarely curative, requiring the development of novel approaches based on innovative molecular targets. In this work, we validated aldehyde dehydrogenase 1A1 and 1A3 isoform expressions in different prostatic tissue-derived cell lines (normal, benign and malignant) and patient-derived primary prostate tumor epithelial cells, demonstrating their potential for therapeutic intervention using a small library of aldehyde dehydrogenase inhibitors.