Quantum Chemical Analysis of Stable Noble Gas Cations for Astrochemical Detection Carlie M
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
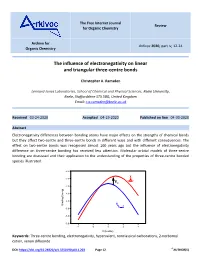
The Influence of Electronegativity on Linear and Triangular Three-Centre Bonds
The Free Internet Journal Review for Organic Chemistry Archive for Arkivoc 2020, part iv, 12-24 Organic Chemistry The influence of electronegativity on linear and triangular three-centre bonds Christopher A. Ramsden Lennard-Jones Laboratories, School of Chemical and Physical Sciences, Keele University, Keele, Staffordshire ST5 5BG, United Kingdom Email: [email protected] Received 03-24-2020 Accepted 04-19-2020 Published on line 04-30-2020 Abstract Electronegativity differences between bonding atoms have major effects on the strengths of chemical bonds but they affect two-centre and three-centre bonds in different ways and with different consequences. The effect on two-centre bonds was recognised almost 100 years ago but the influence of electronegativity difference on three-centre bonding has received less attention. Molecular orbital models of three-centre bonding are discussed and their application to the understanding of the properties of three-centre bonded species illustrated. -2.5 X -3.0 + Y Y Ea -3.5 -4.0 -4.5 X Binding Energy Binding + -5.0 Y Y -5.5 -6.0 -4 -2 0 2 4 h (b units) Keywords: Three-centre bonding, electronegativity, hypervalent, nonclassical carbocations, 2-norbornyl cation, xenon difluoride DOI: https://doi.org/10.24820/ark.5550190.p011.203 Page 12 ©AUTHOR(S) Arkivoc 2020, iv, 12-24 Ramsden, C. A. Table of Contents 1. Introduction 2. Linear Three-Centre Bonds (Hypervalent Bonds) (X-Y-X) X 3. Triangular Three-Centre Bonds (Y Y) 4. The Localised Bond Model of Two- and Three-Centre Bonds 5. Conclusions References -

A Chemical Kinetics Network for Lightning and Life in Planetary Atmospheres P
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by St Andrews Research Repository The Astrophysical Journal Supplement Series, 224:9 (33pp), 2016 May doi:10.3847/0067-0049/224/1/9 © 2016. The American Astronomical Society. All rights reserved. A CHEMICAL KINETICS NETWORK FOR LIGHTNING AND LIFE IN PLANETARY ATMOSPHERES P. B. Rimmer and Ch Helling School of Physics and Astronomy, University of St Andrews, St Andrews, KY16 9SS, UK; [email protected] Received 2015 June 3; accepted 2015 October 22; published 2016 May 23 ABSTRACT There are many open questions about prebiotic chemistry in both planetary and exoplanetary environments. The increasing number of known exoplanets and other ultra-cool, substellar objects has propelled the desire to detect life andprebiotic chemistry outside the solar system. We present an ion–neutral chemical network constructed from scratch, STAND2015, that treats hydrogen, nitrogen, carbon, and oxygen chemistry accurately within a temperature range between 100 and 30,000 K. Formation pathways for glycine and other organic molecules are included. The network is complete up to H6C2N2O3. STAND2015 is successfully tested against atmospheric chemistry models for HD 209458b, Jupiter, and the present-day Earth using a simple one- dimensionalphotochemistry/diffusion code. Our results for the early Earth agree with those of Kasting for CO2,H2, CO, and O2, but do not agree for water and atomic oxygen. We use the network to simulate an experiment where varied chemical initial conditions are irradiated by UV light. The result from our simulation is that more glycine is produced when more ammonia and methane is present. -

Bun)Mo(Pme3)(Η -C2H4) (6) Through Etmgbr Reduction of (Arn)( Bun)Mocl2(DME) in the Presence of Pme3
Synthesis, Catalysis and Stoichiometric Reactivity of Mo Imido Complexes Nicolas A. McLeod BSc. in Chemistry Chemistry Thesis submitted to the Faculty of Graduate Studies In the partial fulfillment of the requirements For the MSc. degree in chemistry Chemistry Department Faculty of Mathematics and Science Brock University St. Catharines, Ontario © Nicolas A. McLeod, 2014 Abstract The current thesis describes the syntheses, catalytic reactivity and mechanistic investigations of novel Mo(IV) and Mo(VI) imido systems. Attempts at preparing mixed bis(imido) Mo(IV) complexes of the type (RN)(R′N)Mo(PMe3)n (n = 2 or 3) derived from the t mono(imido) complexes (RN)Mo(PMe3)3(X)2 (R = Bu (1) or Ar (2); X = Cl2 or HCl, Ar=2,6- i Pr2C6H3) are also described. The addition of lithiated silylamides to 1 or 2 results in the 2 unexpected formation of the C-H activated cyclometallated complexes (RN)Mo(PMe3)2(η - t CH2PMe2)(X) (R = Ar, X = H (3); R = Bu, X = Cl (4)). Complexes 3 and 4 were used in the activation of R′E-H bonds (E = Si, B, C, O, P; R′ = alkyl or aryl), which typically give products of addition across the M-C bond of the type (RN)Mo(PMe3)3(ER′)(X) (4). In the case of 2,6- dimethylphenol, subsequent heating of 4 (R = Ar, R′ = 2,6-Me2C6H3, E = O) to 50 °C results in 2 C-H activation to give the cyclometallated complex (ArN)Mo(PMe3)3(κ -O,C-OPh(Me)CH2) (5). An alternative approach was developed in synthesizing the mixed imido complex t 2 t (ArN)( BuN)Mo(PMe3)(η -C2H4) (6) through EtMgBr reduction of (ArN)( BuN)MoCl2(DME) in the presence of PMe3. -

Preliminary JIRAM Results from Juno Polar Observations: 2. Analysis Of
PUBLICATIONS Geophysical Research Letters RESEARCH LETTER Preliminary JIRAM results from Juno polar observations: 10.1002/2017GL072905 + 2. Analysis of the Jupiter southern H3 emissions Special Section: and comparison with the north aurora Early Results: Juno at Jupiter A. Adriani1 , A. Mura1 , M. L. Moriconi2 , B. M. Dinelli2 , F. Fabiano2,3 , F. Altieri1 , G. Sindoni1 , S. J. Bolton4 , J. E. P. Connerney5 , S. K. Atreya6, F. Bagenal7 , Key Points: + 8 1 1 1 1 1 • H3 intensity, column density, and J.-C. M. C. Gérard , G. Filacchione , F. Tosi , A. Migliorini , D. Grassi , G. Piccioni , temperature maps of the Jupiter R. Noschese1, A. Cicchetti1 , G. R. Gladstone4, C. Hansen9 , W. S. Kurth10 , S. M. Levin11 , southern aurora are derived from B. H. Mauk12 , D. J. McComas13 , A. Olivieri14, D. Turrini1 , S. Stefani1 , and M. Amoroso14 Juno/JIRAM data collected on the first orbit 1INAF-Istituto di Astrofisica e Planetologia Spaziali, Rome, Italy, 2CNR-Istituto di Scienze dell’Atmosfera e del Clima, Bologna, • Emissions from southern aurora are 3 4 more intense than from the North Italy, Dipartimento di Fisica e Astronomia, Università di Bologna, Bologna, Italy, Space Science Department, Southwest • Derived temperatures are in the range Research Institute, San Antonio, Texas, USA, 5Solar System Exploration Division, Planetary Magnetospheres Laboratory, 600°K to 1400°K NASA Goddard Space Flight Center, Greenbelt, Maryland, USA, 6Planetary Science Laboratory, University of Michigan, Ann Arbor, Michigan, USA, 7Laboratory for Atmospheric and Space Physics, University of Colorado Boulder, Boulder, Colorado, USA, 8Laboratoire de Physique Atmosphérique et Planétaire, University of Liège, Liège, Belgium, 9Planetary Science 10 Correspondence to: Institute, Tucson, Arizona, USA, Jet Propulsion Laboratory, California Institute of Technology, Pasadena, California, USA, 11 12 A. -

House Fly Attractants and Arrestante: Screening of Chemicals Possessing Cyanide, Thiocyanate, Or Isothiocyanate Radicals
House Fly Attractants and Arrestante: Screening of Chemicals Possessing Cyanide, Thiocyanate, or Isothiocyanate Radicals Agriculture Handbook No. 403 Agricultural Research Service UNITED STATES DEPARTMENT OF AGRICULTURE Contents Page Methods 1 Results and discussion 3 Thiocyanic acid esters 8 Straight-chain nitriles 10 Propionitrile derivatives 10 Conclusions 24 Summary 25 Literature cited 26 This publication reports research involving pesticides. It does not contain recommendations for their use, nor does it imply that the uses discussed here have been registered. All uses of pesticides must be registered by appropriate State and Federal agencies before they can be recommended. CAUTION: Pesticides can be injurious to humans, domestic animals, desirable plants, and fish or other wildlife—if they are not handled or applied properly. Use all pesticides selectively and carefully. Follow recommended practices for the disposal of surplus pesticides and pesticide containers. ¿/áepé4áaUÁí^a¡eé —' ■ -"" TMK LABIL Mention of a proprietary product in this publication does not constitute a guarantee or warranty by the U.S. Department of Agriculture over other products not mentioned. Washington, D.C. Issued July 1971 For sale by the Superintendent of Documents, U.S. Government Printing Office Washington, D.C. 20402 - Price 25 cents House Fly Attractants and Arrestants: Screening of Chemicals Possessing Cyanide, Thiocyanate, or Isothiocyanate Radicals BY M. S. MAYER, Entomology Research Division, Agricultural Research Service ^ Few chemicals possessing cyanide (-CN), thio- cyanate was slightly attractive to Musca domes- eyanate (-SCN), or isothiocyanate (~NCS) radi- tica, but it was considered to be one of the better cals have been tested as attractants for the house repellents for Phormia regina (Meigen). -

A New Method for the Production of Protonated Hydrogen 29 June 2021
A new method for the production of protonated hydrogen 29 June 2021 + cosmos. This ionized molecule, H 3 (also known as the trihydrogen ion) is made up of three protons and two electrons, and takes the form of an equilateral triangle. Owing to its highly reactive nature, protonated hydrogen promotes the formation of more complex hydrocarbons. It is therefore regarded as an important catalyst for the synthesis of organic, carbon-based molecules that form the basis of life as we know it. + Up until now, H 3 has only been synthesized on Earth from preformed organic compounds or in highly energized hydrogen plasmas. Laser Trihydrogen ions (H+ ) can be formed by subjecting physicists have now discovered a new route for the 3 + water molecules adsorbed to nanoparticles to intense synthesis of H 3 on nanoparticles—in a system that laser light. The experiments mimic the conditions found effectively reproduces conditions under which the in outer space. Credit: Ali Alnaser, AUS molecule can be formed in space. Their findings thus provide new insights into the formation of trihydrogen ions under extraterrestrial conditions. A research group led by Prof. Matthias Kling of the In the experiments, water molecules adsorbed on Max Planck Institute for Quantum Optics (MPQ) the surface of silicon dioxide nanoparticles were and the Ludwig-Maximilian University in Munich irradiated with extremely powerful, ultrashort (LMU), in cooperation with the American University femtosecond laser pulses, essentially mimicking in Sharjah, has discovered a new method for the the effect of the high-energy radiation to which + production of protonated hydrogen (H 3). -

Nuclear Spin Effects in Astrochemistry
Nuclear spin effects in astrochemistry 2-4 May 2017 Grenoble France Table of contents Nuclear spin states in molecules: Unified rotational and permutational symmetry and selection rules in reactive collisions, Hanno Schmiedt [et al.].........4 Spin dynamics of water ice and ortho-to-para ratio of gaseous water desorbed from ice, Tetsuya Hama [et al.]............................6 Preparation, characterization and storage of water vapours highly enriched in its ortho-H2O nuclear spin isomer, P.-A. Turgeon [et al.]................8 Nuclear spin symmetry conservation in H2O investigated by direct absorption FTIR spectroscopy of water vapor cooled down in supersonic expansion, Robert Georges [et al.]..................................... 10 The Ortho-to-Para Ratio in Interstellar Water, Darek Lis............. 12 Spin ratios in comets: complexity of measurements, post-2014 updates, and prospects, Boncho Bonev............................... 14 Nuclear spin restrictions in gas phase reactions: more experiments are needed!, Di- eter Gerlich....................................... 16 Steady-state nuclear spin chemistry in dark clouds, Pierre Hily-Blant....... 18 + Ortho-para transitions of molecular hydrogen in H + H2 collisions, Tomas Gonzalez-Lezana [et al.]................................ 20 Rotational state and ortho-para conversion of H2 on solid surfaces, Katsuyuki Fukutani......................................... 22 H2 ortho-para ratio : the role of dust grains, Emeric Bron [et al.]......... 24 + Ortho/para conversion of H3 in collisions with H2 and H, Octavio Roncero... 26 The ortho:para ratio of the trihydrogen cation in diffuse molecular clouds, Kyle Crabtree......................................... 28 1 What do we know about time scales for the nuclear spin conversion in molecular ices and at the solid-gas interface?, Xavier Michaut [et al.]............. 30 + + Spin states of H2D and D2H as chemical age tracers, Olli Sipil¨a....... -

Molecular Excitation in the Interstellar Medium: Recent Advances In
Molecular excitation in the Interstellar Medium: recent advances in collisional, radiative and chemical processes Evelyne Roueff∗,† and François Lique∗,‡ Laboratoire Univers et Théories, Observatoire de Paris, 92190, Meudon, France, and LOMC - UMR 6294, CNRS-Université du Havre, 25 rue Philippe Lebon, BP 540, 76058, Le Havre, France E-mail: [email protected]; [email protected] Contents 1 Introduction 3 2 Collisional excitation 7 2.1 Methods . 9 2.1.1 Theory . 9 2.1.2 Potential energy surfaces . 10 2.1.3 Scattering Calculations . 13 arXiv:1310.8259v1 [physics.chem-ph] 30 Oct 2013 2.1.4 Experiments . 17 2.2 H2, CO and H2O molecules as benchmark systems . 19 2.2.1 H2 ....................................... 19 ∗To whom correspondence should be addressed †Laboratoire Univers et Théories, Observatoire de Paris, 92190, Meudon, France ‡LOMC - UMR 6294, CNRS-Université du Havre, 25 rue Philippe Lebon, BP 540, 76058, Le Havre, France 1 2.2.2 CO . 22 2.2.3 H2O...................................... 27 2.3 Other recent results . 30 2.3.1 CN / HCN / HNC . 31 2.3.2 CS / SiO / SiS / SO / SO2 .......................... 34 2.3.3 NH3 /NH................................... 38 2.3.4 O2 / OH / NO . 40 2.3.5 C2 /C2H/C3 /C4 / HC3N.......................... 41 2.3.6 Complex Organic Molecules : H2CO / HCOOCH3 / CH3OH . 45 + + + + + 2.3.7 Cations : CH / SiH / HCO /N2H / HOCO . 49 + 2.3.8 H3 ...................................... 52 − − 2.3.9 Anions : CN /C2H ............................ 52 2.4 Isotopologues . 55 3 Radiative and chemical excitation 57 3.1 Radiative effects . 57 3.2 Chemical effects . 59 3.3 Examples . -

Atomic and Molecular Data for State-Resolved Modelling of Hydrogen and Helium and Their Isotopes in Fusion Plasma
INDC(NDS)- 0605 Distr. LP, NE,SK INDC International Nuclear Data Committee Atomic and Molecular Data for State-Resolved Modelling of Hydrogen and Helium and Their Isotopes in Fusion Plasma Summary Report of the First Research Coordination Meeting IAEA Headquarters, Vienna, Austria 10–12 August 2011 Report prepared by B. J. Braams IAEA Nuclear Data Section December 2013 IAEA Nuclear Data Section Vienna International Centre, P.O. Box 100, 1400 Vienna, Austria Selected INDC documents may be downloaded in electronic form from http://www-nds.iaea.org/publications or sent as an e-mail attachment. Requests for hardcopy or e-mail transmittal should be directed to [email protected] or to: Nuclear Data Section International Atomic Energy Agency Vienna International Centre PO Box 100 1400 Vienna Austria Printed by the IAEA in Austria December 2013 INDC(NDS)- 0605 Distr. LP, NE,SK Atomic and Molecular Data for State-Resolved Modelling of Hydrogen and Helium and Their Isotopes in Fusion Plasma Summary Report of the First Research Coordination Meeting IAEA Headquarters, Vienna, Austria 10–12 August 2011 Report prepared by B. J. Braams IAEA Nuclear Data Section Abstract The First Research Coordination Meeting of the IAEA Coordinated Research Project (CRP) on “Atomic and Molecular Data for State-Resolved Modelling of Hydrogen and Helium and Their Isotopes in Fusion Plasma” was held 10-12 August 2011 at IAEA Headquarters in Vienna. Participants reviewed the status of the database on molecular processes of H and He, identified data needs and made plans for development of new data in connection with the CRP. -

An Estimate of the Ortho/Para H2 Ratio Explained by the CO Depletion Theory/Observations
A&A 494, 623–636 (2009) Astronomy DOI: 10.1051/0004-6361:200810587 & c ESO 2009 Astrophysics Chemical modeling of L183 (L134N): an estimate of the ortho/para H2 ratio L. Pagani1, C. Vastel2, E. Hugo3, V. Kokoouline4,C.H.Greene5, A. Bacmann6, E. Bayet7, C. Ceccarelli6,R.Peng8, and S. Schlemmer3 1 LERMA & UMR8112 du CNRS, Observatoire de Paris, 61 Av. de l’Observatoire, 75014 Paris, France e-mail: [email protected] 2 CESR, 9 avenue du colonel Roche, BP 44348, Toulouse Cedex 4, France e-mail: [email protected] 3 I. Physikalisches Institut, Universität zu Köln, Zülpicher Strasse 77, 50937 Köln, Germany e-mail: [hugo;schlemmer]@ph1.uni-koeln.de 4 Department of Physics, University of Central Florida, Orlando, 32816, USA e-mail: [email protected] 5 Department of Physics and JILA, University of Colorado, Boulder, Colorado 80309-0440, USA e-mail: [email protected] 6 Laboratoire d’Astrophysique de Grenoble, Université Joseph Fourier, UMR 5571 du CNRS, 414 rue de la Piscine, 38041 Grenoble Cedex 09, France e-mail: [cecilia.ceccarelli;aurore.bacmann]@obs.ujf-grenoble.fr 7 Department of physics and astronomy, University College London, Gower street, London WC1E 6BT, UK e-mail: [email protected] 8 Caltech Submillimeter Observatory, 111 Nowelo Street, Hilo, HI 96720, USA e-mail: [email protected] Received 14 July 2008 / Accepted 6 October 2008 ABSTRACT + Context. The high degree of deuteration observed in some prestellar cores depends on the ortho-to-para H2 ratio through the H3 fractionation. Aims. We want to constrain the ortho/para H2 ratio across the L183 prestellar core. -
![Arxiv:1510.07052V1 [Astro-Ph.EP] 23 Oct 2015 99 Lae Ta.20,Adrfrne Therein)](https://docslib.b-cdn.net/cover/7858/arxiv-1510-07052v1-astro-ph-ep-23-oct-2015-99-lae-ta-20-adrfrne-therein-2397858.webp)
Arxiv:1510.07052V1 [Astro-Ph.EP] 23 Oct 2015 99 Lae Ta.20,Adrfrne Therein)
A Chemical Kinetics Network for Lightning and Life in Planetary Atmospheres P. B. Rimmer1 and Ch Helling School of Physics and Astronomy, University of St Andrews, St Andrews, KY16 9SS, United Kingdom ABSTRACT There are many open questions about prebiotic chemistry in both planetary and exoplane- tary environments. The increasing number of known exoplanets and other ultra-cool, substellar objects has propelled the desire to detect life and prebiotic chemistry outside the solar system. We present an ion-neutral chemical network constructed from scratch, Stand2015, that treats hydrogen, nitrogen, carbon and oxygen chemistry accurately within a temperature range between 100 K and 30000 K. Formation pathways for glycine and other organic molecules are included. The network is complete up to H6C2N2O3. Stand2015 is successfully tested against atmo- spheric chemistry models for HD209458b, Jupiter and the present-day Earth using a simple 1D photochemistry/diffusion code. Our results for the early Earth agree with those of Kasting (1993) for CO2, H2, CO and O2, but do not agree for water and atomic oxygen. We use the network to simulate an experiment where varied chemical initial conditions are irradiated by UV light. The result from our simulation is that more glycine is produced when more ammonia and methane is present. Very little glycine is produced in the absence of any molecular nitrogen and oxygen. This suggests that production of glycine is inhibited if a gas is too strongly reducing. Possible applications and limitations of the chemical kinetics network are also discussed. Subject headings: astrobiology — atmospheric effects — molecular processes — planetary systems 1. Introduction The input energy source and the initial chem- istry have been varied across these different ex- The potential connection between a focused periments. -

George Andrew Olah Across Conventional Lines
GENERAL ARTICLE George Andrew Olah Across Conventional Lines Ripudaman Malhotra, Thomas Mathew and G K Surya Prakash Hungarian born American chemist, George Andrew Olah was aprolific researcher. The central theme of his career was the 12 pursuit of structure and mechanisms in chemistry, particu- larly focused on electron-deficient intermediates. He leaves behind a large body of work comprising almost 1500 papers and twenty books for the scientific community. Some selected works have been published in three aptly entitled volumes, 3 Across Conventional Lines. There is no way to capture the many contributions of Olah in a short essay. For this appreciation, we have chosen to high- 1 light some of those contributions that to our mind represent Ripudaman Malhotra, a student of Prof. Olah, is a asignificant advance to the state of knowledge. retired chemist who spent his entire career at SRI International working mostly Organofluorine Chemistry on energy-related issues. He co-authored the book on Early on in his career, while still in Hungary, Olah began studying global energy – A Cubic Mile organofluorine compounds. Olah’s interest in fluorinated com- of Oil. pounds was piqued by the theoretical ramifications of a strong 2 Thomas Mathew, a senior C-F bond. Thus, whereas chloromethanol immediately decom- scientist at the Loker Hydrocarbon Research poses into formaldehyde and HCl, he reasoned that the stronger Institute, has been a close C-F bond might render fluoromethanol stable. He succeeded in associate of Prof. Olah over preparing fluoromethanol by the reduction of ethyl flouorofor- two decades. 3 mate with lithium aluminum hydride [1].