Wednesday, March 13, 2019 4:00Pm
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

DRUGS REQUIRING PRIOR AUTHORIZATION in the MEDICAL BENEFIT Page 1
Effective Date: 08/01/2021 DRUGS REQUIRING PRIOR AUTHORIZATION IN THE MEDICAL BENEFIT Page 1 Therapeutic Category Drug Class Trade Name Generic Name HCPCS Procedure Code HCPCS Procedure Code Description Anti-infectives Antiretrovirals, HIV CABENUVA cabotegravir-rilpivirine C9077 Injection, cabotegravir and rilpivirine, 2mg/3mg Antithrombotic Agents von Willebrand Factor-Directed Antibody CABLIVI caplacizumab-yhdp C9047 Injection, caplacizumab-yhdp, 1 mg Cardiology Antilipemic EVKEEZA evinacumab-dgnb C9079 Injection, evinacumab-dgnb, 5 mg Cardiology Hemostatic Agent BERINERT c1 esterase J0597 Injection, C1 esterase inhibitor (human), Berinert, 10 units Cardiology Hemostatic Agent CINRYZE c1 esterase J0598 Injection, C1 esterase inhibitor (human), Cinryze, 10 units Cardiology Hemostatic Agent FIRAZYR icatibant J1744 Injection, icatibant, 1 mg Cardiology Hemostatic Agent HAEGARDA c1 esterase J0599 Injection, C1 esterase inhibitor (human), (Haegarda), 10 units Cardiology Hemostatic Agent ICATIBANT (generic) icatibant J1744 Injection, icatibant, 1 mg Cardiology Hemostatic Agent KALBITOR ecallantide J1290 Injection, ecallantide, 1 mg Cardiology Hemostatic Agent RUCONEST c1 esterase J0596 Injection, C1 esterase inhibitor (recombinant), Ruconest, 10 units Injection, lanadelumab-flyo, 1 mg (code may be used for Medicare when drug administered under Cardiology Hemostatic Agent TAKHZYRO lanadelumab-flyo J0593 direct supervision of a physician, not for use when drug is self-administered) Cardiology Pulmonary Arterial Hypertension EPOPROSTENOL (generic) -

Denosumab Versus Romosozumab for Postmenopausal Osteoporosis Treatment
www.nature.com/scientificreports OPEN Denosumab versus romosozumab for postmenopausal osteoporosis treatment Tomonori Kobayakawa1, Akiko Miyazaki2, Makoto Saito3, Takako Suzuki2,4, Jun Takahashi2 & Yukio Nakamura2* Denosumab and romosozumab, a recently approved new drug, are efective and widely known molecular-targeted drugs for postmenopausal osteoporosis treatment. However, no studies have directly compared their therapeutic efects or safety in postmenopausal osteoporosis. This retrospective observational registry study compared the efcacy of 12-month denosumab or romosozumab treatment in postmenopausal osteoporosis patients. The primary outcome was the change in bone mineral density (BMD) at the lumbar spine. Secondary outcomes included BMD changes at the total hip and femoral neck, changes in bone turnover markers, and adverse events. Propensity score matching was employed to assemble patient groups with similar baseline characteristics. Sixty-nine patients each received either denosumab or romosozumab for 12 months. The mean 12-month percentage change from baseline in lumbar spine BMD was 7.2% in the denosumab group and 12.5% in the romosozumab group, indicating a signifcant diference between the groups. The percentage changes in BMD at both the total hip and femoral neck were also signifcantly higher at 12 months in the romosozumab group than in the denosumab group. In denosumab patients, bone formation and bone resorption markers were signifcantly decreased at 6 and 12 months from baseline. In the romosozumab group, the bone formation marker was signifcantly increased at 6 months and then returned to baseline, while the bone resorption marker was signifcantly decreased at both time points. Adverse events were few and predominantly minor in both groups, with no remarkable diference in the incidence of new vertebral fractures. -

Two Phase 3 Clinical Trials Comparing the Safety and Efficacy of Netarsudil to Timolol in Patients with Elevated Intraocular
Two Phase 3 Clinical Trials Comparing the Safety and Efficacy of Netarsudil to Timolol in Patients With Elevated Intraocular Pressure: Rho Kinase Elevated IOP Treatment Trial 1 and 2 (ROCKET-1 and ROCKET-2) JANET B. SERLE, L. JAY KATZ, EUGENE MCLAURIN, THERESA HEAH, NANCY RAMIREZ-DAVIS, DALE W. USNER, GARY D. NOVACK, AND CASEY C. KOPCZYNSKI, FOR THE ROCKET-1 AND ROCKET-2 STUDY GROUPS PURPOSE: To evaluate the efficacy and ocular and sys- ROCKET-2) for timolol (P < .0001 for netarsudil vs temic safety of netarsudil 0.02% ophthalmic solution, a timolol). rho-kinase inhibitor and norepinephrine transporter in- CONCLUSIONS: In 2 large, randomized, double-masked hibitor, in patients with open-angle glaucoma and ocular trials reported here, once-daily dosing of netarsudil hypertension. 0.02% was found to be effective and well tolerated for DESIGN: Double-masked, randomized noninferiority the treatment of patients with ocular hypertension and clinical trials: Rho Kinase Elevated IOP Treatment Trial open-angle glaucoma. The novel pharmacology and 1 and 2 (ROCKET-1 and ROCKET-2). aqueous humor dynamic effects of this molecule suggest METHODS: After a washout of all pre-study ocular hy- it may be a useful addition to the armamentarium of potensive medications, eligible patients were randomized ocular hypotensive medications. (Am J Ophthalmol to receive netarsudil 0.02% once daily (q.d.), timolol 2018;186:116–127. Ó 2017 The Authors. Published 0.5% twice a day (b.i.d.), and (ROCKET-2 only) netar- by Elsevier Inc. This is an open access article under the sudil 0.02% b.i.d. -

[Product Monograph Template
PRODUCT MONOGRAPH INCLUDING PATIENT MEDICATION INFORMATION Kinevac® (Sincalide) For Injection 5 mcg/vial Diagnostic Cholecystokinetic Bracco Imaging Canada Date of Initial Approval: Montreal, Quebec July 2, 1996 Canada, H1J 2Z4 Date of Revision: October 30, 2019 Submission Control No: 231066 Product Monograph Kinevac® Page 1 of 14 TABLE OF CONTENTS TABLE OF CONTENTS ............................................................................................................. 2 PART I: HEALTH PROFESSIONAL INFORMATION ................................................................. 3 1 INDICATIONS ................................................................................................................. 3 Pediatrics ........................................................................................................................ 3 Geriatrics ......................................................................................................................... 3 2 CONTRAINDICATIONS .................................................................................................. 3 3 DOSAGE AND ADMINISTRATION ................................................................................ 4 Recommended Dose and Dosage Adjustment ................................................................ 4 Reconstitution .................................................................................................................. 4 4 OVERDOSAGE .............................................................................................................. -

Increased Bone Mineral Density for 1 Year of Romosozumab, Vs Placebo, Followed by 2 Years of Denosumab in the Japanese Subgroup
Archives of Osteoporosis (2019) 14:59 https://doi.org/10.1007/s11657-019-0608-z ORIGINAL ARTICLE Increased bone mineral density for 1 year of romosozumab, vs placebo, followed by 2 years of denosumab in the Japanese subgroup of the pivotal FRAME trial and extension Akimitsu Miyauchi1 & Rajani V. Dinavahi2 & Daria B. Crittenden2 & Wenjing Yang2 & Judy C. Maddox2 & Etsuro Hamaya3 & Yoichi Nakamura3 & Cesar Libanati4 & Andreas Grauer2 & Junichiro Shimauchi3 Received: 1 March 2019 /Accepted: 18 May 2019 # The Author(s) 2019 Abstract Summary Romosozumab, which binds sclerostin, rebuilds the skeletal foundation before transitioning to antiresorptive treat- ment. This subgroup analysis of an international, randomized, double-blind study in postmenopausal women with osteoporosis showed efficacy and safety outcomes for romosozumab followed by denosumab in Japanese women were generally consistent with those for the overall population. Purpose In the international, randomized, double-blind, phase 3 FRActure study, in postmenopausal woMen with ostEoporosis (FRAME; NCT01575834), romosozumab followed by denosumab significantly improved bone mineral density (BMD) and reduced fracture risk. This report evaluates Japanese women in FRAME. Methods Postmenopausal women with osteoporosis (T-score − 3.5 to − 2.5 at total hip or femoral neck) received romosozumab 210 mg or placebo subcutaneously monthly for 12 months, then each group received denosumab 60 mg subcutaneously every 6 months for 24 months. The key endpoint for Japanese women was BMD change. Other endpoints included new vertebral, clinical, and nonvertebral fracture; the subgroup analysis did not have adequate power to demonstrate statistically significant reductions. Results Of 7180 enrolled subjects, 492 (6.9%) were Japanese (247 romosozumab, 245 placebo). -

Table 1. Glaucoma Medications: Mechanisms, Dosing and Precautions Brand Generic Mechanism of Action Dosage/Avg
OPTOMETRIC STUDY CENTER Table 1. Glaucoma Medications: Mechanisms, Dosing and Precautions Brand Generic Mechanism of Action Dosage/Avg. % Product Sizes Side Effects Warnings Reduction CHOLINERGIC AGENTS Direct Pilocarpine (generic) Pilocarpine 1%, 2%, 4% Increases trabecular outflow BID-QID/15-25% 15ml Headache, blurred vision, myopia, retinal detachment, bronchiole constriction, Angle closure, shortness of breath, retinal narrowing of angle detachment Indirect Phospholine Iodide (Pfizer) Echothiophate iodide 0.125% Increases trabecular outflow QD-BID/15-25% 5ml Same as above plus cataractogenic iris cysts in children, pupillary block, Same as above, plus avoid prior to any increased paralysis with succinylcholine general anesthetic procedure ALPHA-2 AGONISTS Alphagan P (Allergan) Brimonidine tartrate 0.1%, 0.15% with Purite Decreases aqueous production, increases BID-TID/up to 26% 5ml, 10ml, 15ml Dry mouth, hypotension, bradycardia, follicular conjunctivitis, ocular irritation, Monitor for shortness of breath, dizziness, preservative uveoscleral outflow pruritus, dermatitis, conjunctival blanching, eyelid retraction, mydriasis, drug ocular redness and itching, fatigue allergy Brimonidine tartrate Brimonidine tartrate 0.15%, 0.2% Same as above Same as above 5ml, 10ml Same as above Same as above (generic) Iopidine (Novartis) Apraclonidine 0.5% Decreases aqueous production BID-TID/up to 25% 5ml, 10ml Same as above but higher drug allergy (40%) Same as above BETA-BLOCKERS Non-selective Betagan (Allergan) Levobunolol 0.25%, 0.5% Decreases -

Rhopressa™ Netarsudil Ophthalmic Solution 0.02%
Rhopressa™ Netarsudil ophthalmic solution 0.02% CDER Dermatologic and Ophthalmic Drugs Advisory Committee October 13, 2017 Aerie Pharmaceuticals, Inc. 1 Introduction Marvin Garrett Vice President, Regulatory Affairs and Quality Assurance Aerie Pharmaceuticals, Inc. 2 Aerie Pharmaceuticals • 2005: Aerie founded as a spin-out from Duke University: – Dr. Eric Toone – Dr. Casey Kopczynski – Dr. David Epstein – Dr. Epstein’s goal from the beginning: Develop a therapy that targeted the diseased tissue in glaucoma, the trabecular outflow pathway • 2006: Aerie discovered its first Rho kinase inhibitor • 2009: Aerie invented netarsudil • 2012: Netarsudil 1st clinical study • 2017: NDA filed 3 Netarsudil: A New Drug Class for Lowering IOP We are requesting a recommendation for approval of netarsudil ophthalmic solution 0.02% for reduction of intraocular pressure (IOP) in patients with open-angle glaucoma or ocular hypertension given one drop QD 4 Agenda Unmet Medical Needs Richard A. Lewis, MD Chief Medical Officer Aerie Pharmaceuticals, Inc. Past President, American Glaucoma Society Program Design and Efficacy Casey Kopczynski, PhD Chief Scientific Officer Aerie Pharmaceuticals, Inc. Safety Theresa Heah, MD, MBA VP Clinical Research and Medical Affairs Aerie Pharmaceuticals, Inc. Benefits and Risks Janet Serle, MD Professor of Ophthalmology Glaucoma Fellowship Director Icahn School of Medicine at Mount Sinai 5 List of Expert Responders • Cynthia Mattox, MD – Associate Professor of Ophthalmology, Tufts University School of Medicine – Current President, American Glaucoma Society • Mark Reasor, PhD – Professor of Physiology & Pharmacology, Robert C. Byrd Health Sciences Center, West Virginia University • Bennie H. Jeng, MD – Professor and Chair, Department of Ophthalmology & Visual Sciences, University of Maryland School of Medicine • Dale Usner, PhD – Biostatistics Consultant to Aerie Pharmaceuticals, Inc. -

Denosumab, Raloxifene, Romosozumab and Teriparatide to Prevent Osteoporotic Fragility Fractures: a Systematic Review and Economic Evaluation
Journals Library Health Technology Assessment Volume 24 • Issue 29 • June 2020 ISSN 1366-5278 Denosumab, raloxifene, romosozumab and teriparatide to prevent osteoporotic fragility fractures: a systematic review and economic evaluation Sarah Davis, Emma Simpson, Jean Hamilton, Marrissa Martyn-St James, Andrew Rawdin, Ruth Wong, Edward Goka, Neil Gittoes and Peter Selby DOI 10.3310/hta24290 Denosumab, raloxifene, romosozumab and teriparatide to prevent osteoporotic fragility fractures: a systematic review and economic evaluation Sarah Davis ,1* Emma Simpson ,1 Jean Hamilton ,1 Marrissa Martyn-St James ,1 Andrew Rawdin ,1 Ruth Wong ,1 Edward Goka ,1 Neil Gittoes 2 and Peter Selby 3 1Health Economics and Decision Science, School of Health and Related Research (ScHARR), University of Sheffield, Sheffield, UK 2University Hospitals Birmingham NHS Foundation Trust, Birmingham, UK 3School of Medical Sciences, University of Manchester, Manchester, UK *Corresponding author Declared competing interests of authors: Neil Gittoes reports personal fees for being a member of the advisory board to Union Chimique Belge (UCB) S.A. (Brussels, Belgium) and personal fees for contributing to educational meeting sponsored by Eli Lilly and Company (Indianapolis, IN, USA), outside the submitted work. He is also a trustee of the National Osteoporosis Society, a member of the advisory board of the National Osteoporosis Guideline Group and Deputy Chairperson of the Specialised Endocrinology Clinical Reference Group, NHS England. Published June 2020 DOI: 10.3310/hta24290 This report should be referenced as follows: Davis S, Simpson E, Hamilton J, James MM-S, Rawdin A, Wong R, et al. Denosumab, raloxifene, romosozumab and teriparatide to prevent osteoporotic fragility fractures: a systematic review and economic evaluation. -

Looking for New Classes of Bronchodilators
REVIEW BRONCHODILATORS The future of bronchodilation: looking for new classes of bronchodilators Mario Cazzola1, Paola Rogliani 1 and Maria Gabriella Matera2 Affiliations: 1Dept of Experimental Medicine, University of Rome Tor Vergata, Rome, Italy. 2Dept of Experimental Medicine, University of Campania “Luigi Vanvitelli”, Naples, Italy. Correspondence: Mario Cazzola, Dept of Experimental Medicine, University of Rome Tor Vergata, Via Montpellier 1, Rome, 00133, Italy. E-mail: [email protected] @ERSpublications There is a real interest among researchers and the pharmaceutical industry in developing novel bronchodilators. There are several new opportunities; however, they are mostly in a preclinical phase. They could better optimise bronchodilation. http://bit.ly/2lW1q39 Cite this article as: Cazzola M, Rogliani P, Matera MG. The future of bronchodilation: looking for new classes of bronchodilators. Eur Respir Rev 2019; 28: 190095 [https://doi.org/10.1183/16000617.0095-2019]. ABSTRACT Available bronchodilators can satisfy many of the needs of patients suffering from airway disorders, but they often do not relieve symptoms and their long-term use raises safety concerns. Therefore, there is interest in developing new classes that could help to overcome the limits that characterise the existing classes. At least nine potential new classes of bronchodilators have been identified: 1) selective phosphodiesterase inhibitors; 2) bitter-taste receptor agonists; 3) E-prostanoid receptor 4 agonists; 4) Rho kinase inhibitors; 5) calcilytics; 6) agonists of peroxisome proliferator-activated receptor-γ; 7) agonists of relaxin receptor 1; 8) soluble guanylyl cyclase activators; and 9) pepducins. They are under consideration, but they are mostly in a preclinical phase and, consequently, we still do not know which classes will actually be developed for clinical use and whether it will be proven that a possible clinical benefit outweighs the impact of any adverse effect. -

Title 16. Crimes and Offenses Chapter 13. Controlled Substances Article 1
TITLE 16. CRIMES AND OFFENSES CHAPTER 13. CONTROLLED SUBSTANCES ARTICLE 1. GENERAL PROVISIONS § 16-13-1. Drug related objects (a) As used in this Code section, the term: (1) "Controlled substance" shall have the same meaning as defined in Article 2 of this chapter, relating to controlled substances. For the purposes of this Code section, the term "controlled substance" shall include marijuana as defined by paragraph (16) of Code Section 16-13-21. (2) "Dangerous drug" shall have the same meaning as defined in Article 3 of this chapter, relating to dangerous drugs. (3) "Drug related object" means any machine, instrument, tool, equipment, contrivance, or device which an average person would reasonably conclude is intended to be used for one or more of the following purposes: (A) To introduce into the human body any dangerous drug or controlled substance under circumstances in violation of the laws of this state; (B) To enhance the effect on the human body of any dangerous drug or controlled substance under circumstances in violation of the laws of this state; (C) To conceal any quantity of any dangerous drug or controlled substance under circumstances in violation of the laws of this state; or (D) To test the strength, effectiveness, or purity of any dangerous drug or controlled substance under circumstances in violation of the laws of this state. (4) "Knowingly" means having general knowledge that a machine, instrument, tool, item of equipment, contrivance, or device is a drug related object or having reasonable grounds to believe that any such object is or may, to an average person, appear to be a drug related object. -

PDCO Monthly Report of Opinions on Paediatric Investigation Plans and Other Activities 13-16 October 2020
16 October 2020 EMA/578758/2020 Human Medicines Division PDCO monthly report of opinions on paediatric investigation plans and other activities 13-16 October 2020 Opinions on paediatric investigation plans The Paediatric Committee (PDCO) adopted opinions agreeing paediatric investigation plans (PIPs) for the following medicines: • Bimekizumab, EMEA-002189-PIP03-19, from UCB Biopharma SRL, for the treatment of chronic idiopathic arthritis (including rheumatoid arthritis, spondyloarthritis, psoriatic arthritis and juvenile idiopathic arthritis); • Adrenaline (epinephrine), EMEA-002749-PIP01-19, from ARS Pharmaceuticals IRL, Limited, for the treatment of allergic reactions; • Guselkumab, EMEA-001523-PIP04-19, from Janssen-Cilag International N.V., for the treatment of ulcerative colitis; • Dapirolizumab pegol, EMEA-002702-PIP01-19, from UCB Biopharma SRL, for the treatment of systemic lupus erythematosus; • Allogeneic bone marrow derived mesenchymal stromal cells, ex-vivo expanded (MC0518), EMEA- 002706-PIP01-19, from medac Gesellschaft für klinische Spezialpräparate mbH, for the treatment of acute graft-versus-host disease; • Alpha1-proteinase inhibitor (human), EMEA-001312-PIP03-19, from CSL Behring GmbH, for the treatment of acute graft-versus-host disease; • Adeno-associated viral vector serotype 9 containing the human mini-dystrophin gene (PF- 06939926), EMEA-002741-PIP01-20, from Pfizer Europe MA EEIG, for the treatment of Duchenne Muscular Dystrophy; • Venglustat, EMEA-001716-PIP04-19, from Genzyme Europe B.V., for the treatment of -
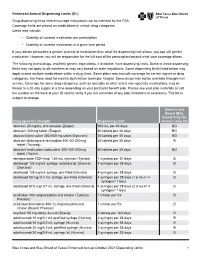
Enhanced Annual Drug List Dispensing Limits
Enhanced Annual Dispensing Limits (DL) Drug dispensing limits help encourage medication use as intended by the FDA. Coverage limits are placed on medications in certain drug categories. Limits may include: • Quantity of covered medication per prescription • Quantity of covered medication in a given time period If your doctor prescribes a greater quantity of medication than what the dispensing limit allows, you can still get the medication. However, you will be responsible for the full cost of the prescription beyond what your coverage allows. The following brand drugs, and their generic equivalents, if available, have dispensing limits. Some of these dispensing limits may not apply to all members or may vary based on state regulations. Some dispensing limits listed below may apply across multiple medications within a drug class. Some plans may exclude coverage for certain agents or drug categories, like those used for erectile dysfunction (example: Viagra). Some drugs may not be available through mail service. Coverage for some drug categories, such as specialty or other select non‑specialty medications, may be limited to a 30‑day supply at a time depending on your particular benefit plan. Please see your plan materials or call the number on the back of your ID card to verify if you are uncertain of any plan limitations or exclusions. This list is subject to change. Generic and Brand (BG), Brand Only (B), Drug (generic) strength Dispensing Limit Generic only (G) abacavir 20 mg/mL oral solution (Ziagen) 960 mL per 30 days BG abacavir 300