Leukoencephalopathy with Accumulated Succinate Is Indicative
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Orphan Disease Networks
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector ARTICLE The Orphan Disease Networks Minlu Zhang,1,3,5 Cheng Zhu,1,5 Alexis Jacomy,4 Long J. Lu,1,2,3 and Anil G. Jegga1,2,3,* The low prevalence rate of orphan diseases (OD) requires special combined efforts to improve diagnosis, prevention, and discovery of novel therapeutic strategies. To identify and investigate relationships based on shared genes or shared functional features, we have con- ducted a bioinformatic-based global analysis of all orphan diseases with known disease-causing mutant genes. Starting with a bipartite network of known OD and OD-causing mutant genes and using the human protein interactome, we first construct and topologically analyze three networks: the orphan disease network, the orphan disease-causing mutant gene network, and the orphan disease-causing mutant gene interactome. Our results demonstrate that in contrast to the common disease-causing mutant genes that are predomi- nantly nonessential, a majority of orphan disease-causing mutant genes are essential. In confirmation of this finding, we found that OD-causing mutant genes are topologically important in the protein interactome and are ubiquitously expressed. Additionally, func- tional enrichment analysis of those genes in which mutations cause ODs shows that a majority result in premature death or are lethal in the orthologous mouse gene knockout models. To address the limitations of traditional gene-based disease networks, we also construct and analyze OD networks on the basis of shared enriched features (biological processes, cellular components, pathways, phenotypes, and literature citations). -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Mutations in SDHD Lead to Autosomal Recessive Encephalomyopathy And
Downloaded from http://jmg.bmj.com/ on December 5, 2017 - Published by group.bmj.com Genotype-phenotype correlations ORIGINAL ARTICLE Mutations in SDHD lead to autosomal recessive encephalomyopathy and isolated mitochondrial complex II deficiency Christopher Benjamin Jackson,1,2 Jean-Marc Nuoffer,3 Dagmar Hahn,3 Holger Prokisch,4,5 Birgit Haberberger,4 Matthias Gautschi,3,6 Annemarie Häberli,3 Sabina Gallati,1 André Schaller1 ▸ Additional material is ABSTRACT succinate to fumarate, is formed by the two larger published online only. To view Background Defects of the mitochondrial respiratory hydrophilic subunits, SDHA and SDHB, which also please visit the journal online (10.1136/jmedgenet-2013- chain complex II (succinate dehydrogenase (SDH) harbour the redox cofactors that participate in elec- 101932) complex) are extremely rare. Of the four nuclear encoded tron transfer to ubiquinone. The cofactor FAD is 1 proteins composing complex II, only mutations in the covalently bound to SDHA which provides the suc- Division of Human Genetics, fl fi Departments of Paediatrics and 70 kDa avoprotein (SDHA) and the recently identi ed cinate binding site, and SDHB possesses three Fe-S Clinical Research, University of complex II assembly factor (SDHAF1) have been found to centres which mediate the electron transfer to ubi- Bern, Bern, Switzerland be causative for mitochondrial respiratory chain diseases. quinone. The smaller hydrophobic SDHC and 2 Graduate School for Cellular Mutations in the other three subunits (SDHB, SDHC, SDHD subunits constitute the membrane anchor and Biomedical Sciences, SDHD) and the second assembly factor (SDHAF2) have and ubiquinone binding sites of CII and localise University of Bern, Bern, 2–4 Switzerland so far only been associated with hereditary CII to the inner mitochondrial membrane. -

Crystal Structure of Bacterial Succinate:Quinone Oxidoreductase Flavoprotein Sdha in Complex with Its Assembly Factor Sdhe
Crystal structure of bacterial succinate:quinone oxidoreductase flavoprotein SdhA in complex with its assembly factor SdhE Megan J. Mahera,1, Anuradha S. Heratha, Saumya R. Udagedaraa, David A. Dougana, and Kaye N. Truscotta,1 aDepartment of Biochemistry and Genetics, La Trobe Institute for Molecular Science, La Trobe University, Melbourne, VIC 3086, Australia Edited by Amy C. Rosenzweig, Northwestern University, Evanston, IL, and approved February 14, 2018 (received for review January 4, 2018) Succinate:quinone oxidoreductase (SQR) functions in energy me- quinol:FRD), respectively (13, 15). The importance of this protein tabolism, coupling the tricarboxylic acid cycle and electron transport family, in normal cellular metabolism, is manifested by the iden- chain in bacteria and mitochondria. The biogenesis of flavinylated tification of a mutation in human SDHAF2 (Gly78Arg), which is SdhA, the catalytic subunit of SQR, is assisted by a highly conserved linked to an inherited neuroendocrine disorder, PGL2 (10). Cur- assembly factor termed SdhE in bacteria via an unknown mecha- rently, however, the role of SdhE in flavinylation remains poorly nism. By using X-ray crystallography, we have solved the structure understood. To date, three different modes of action for SdhE/ of Escherichia coli SdhE in complex with SdhA to 2.15-Å resolution. Sdh5 have been proposed, suggesting that SdhE facilitates the Our structure shows that SdhE makes a direct interaction with the binding and delivery of FAD (13), acts as a chaperone for SdhA flavin adenine dinucleotide-linked residue His45 in SdhA and main- (10), or catalyzes the attachment of FAD (10). Moreover, the re- tains the capping domain of SdhA in an “open” conformation. -

Mitofusins Regulate Lipid Metabolism to Mediate the Development of Lung Fibrosis
ARTICLE https://doi.org/10.1038/s41467-019-11327-1 OPEN Mitofusins regulate lipid metabolism to mediate the development of lung fibrosis Kuei-Pin Chung 1,2,3, Chia-Lang Hsu 4, Li-Chao Fan1, Ziling Huang1, Divya Bhatia 5, Yi-Jung Chen6, Shu Hisata1, Soo Jung Cho1, Kiichi Nakahira1, Mitsuru Imamura 1, Mary E. Choi5,7, Chong-Jen Yu5,8, Suzanne M. Cloonan1 & Augustine M.K. Choi1,7 Accumulating evidence illustrates a fundamental role for mitochondria in lung alveolar type 2 1234567890():,; epithelial cell (AEC2) dysfunction in the pathogenesis of idiopathic pulmonary fibrosis. However, the role of mitochondrial fusion in AEC2 function and lung fibrosis development remains unknown. Here we report that the absence of the mitochondrial fusion proteins mitofusin1 (MFN1) and mitofusin2 (MFN2) in murine AEC2 cells leads to morbidity and mortality associated with spontaneous lung fibrosis. We uncover a crucial role for MFN1 and MFN2 in the production of surfactant lipids with MFN1 and MFN2 regulating the synthesis of phospholipids and cholesterol in AEC2 cells. Loss of MFN1, MFN2 or inhibiting lipid synthesis via fatty acid synthase deficiency in AEC2 cells exacerbates bleomycin-induced lung fibrosis. We propose a tenet that mitochondrial fusion and lipid metabolism are tightly linked to regulate AEC2 cell injury and subsequent fibrotic remodeling in the lung. 1 Division of Pulmonary and Critical Care Medicine, Joan and Sanford I. Weill Department of Medicine, Weill Cornell Medicine, New York, NY 10021, USA. 2 Department of Laboratory Medicine, National Taiwan University Hospital and National Taiwan University Cancer Center, Taipei 10002, Taiwan. 3 Graduate Institute of Clinical Medicine, College of Medicine, National Taiwan University, Taipei 10051, Taiwan. -

Succinate Dehydrogenase Inhibition Leads to Epithelial-Mesenchymal Transition and Reprogrammed Carbon Metabolism." Cancer & Metabolism
Succinate dehydrogenase inhibition leads to epithelial- mesenchymal transition and reprogrammed carbon metabolism The MIT Faculty has made this article openly available. Please share how this access benefits you. Your story matters. Citation Aspuria, Paul-Joseph P., Sophia Y. Lunt, Leif Varemo, Laurent Vergnes et al. "Succinate dehydrogenase inhibition leads to epithelial-mesenchymal transition and reprogrammed carbon metabolism." Cancer & Metabolism. (2014 Dec 15); 2 (1):21. As Published http://dx.doi.org/10.1186/2049-3002-2-21 Publisher BioMed Central Ltd. Version Final published version Citable link http://hdl.handle.net/1721.1/92499 Terms of Use Creative Commons Attribution Detailed Terms http://creativecommons.org/licenses/by/4.0 Aspuria et al. Cancer & Metabolism 2014, 2:21 http://www.cancerandmetabolism.com/content/2/1/21 Cancer & Metabolism RESEARCH Open Access Succinate dehydrogenase inhibition leads to epithelial-mesenchymal transition and reprogrammed carbon metabolism Paul-Joseph P Aspuria1*, Sophia Y Lunt2,3†, Leif Väremo4†, Laurent Vergnes5, Maricel Gozo1,6, Jessica A Beach1,6, Brenda Salumbides1,KarenReue5, W Ruprecht Wiedemeyer1,7,JensNielsen4, Beth Y Karlan1,7 and Sandra Orsulic1,7 Abstract Background: Succinate dehydrogenase (SDH) is a mitochondrial metabolic enzyme complex involved in both the electron transport chain and the citric acid cycle. SDH mutations resulting in enzymatic dysfunction have been found to be a predisposing factor in various hereditary cancers. Therefore, SDH has been implicated as a tumor suppressor. Results: We identified that dysregulation of SDH components also occurs in serous ovarian cancer, particularly the SDH subunit SDHB. Targeted knockdown of Sdhb in mouse ovarian cancer cells resulted in enhanced proliferation and an epithelial-to-mesenchymal transition (EMT). -

Systems Genetics Analysis of the LXS Recombinant Inbred Mouse Strains:Genetic and Molecular Insights Into Acute Ethanol Tolerance
PLOS ONE RESEARCH ARTICLE Systems genetics analysis of the LXS recombinant inbred mouse strains:Genetic and molecular insights into acute ethanol tolerance 1,2 3,4,5 3 3 Richard A. RadcliffeID *, Robin DowellID , Aaron T. Odell , Phillip A. RichmondID , 1 1 6 1 Beth Bennett , Colin LarsonID , Katerina KechrisID , Laura M. SabaID , 7 6 Pratyaydipta RudraID , Shi WenID a1111111111 1 Skaggs School of Pharmacy and Pharmaceutical Sciences, University of Colorado Anschutz Medical Campus, Aurora, CO, United States of America, 2 Institute for Behavioral Genetics, University of Colorado a1111111111 Boulder, Boulder CO, United States of America, 3 BioFrontiers Institute, University of Colorado Boulder, a1111111111 Boulder, CO, United States of America, 4 Department of Molecular, Cellular, and Developmental Biology, a1111111111 University of Colorado Boulder, Boulder, CO, United States of America, 5 Department of Computer Science, a1111111111 University of Colorado Boulder, Boulder, CO, United States of America, 6 Department of Biostatistics and Informatics, Colorado School of Public Health, University of Colorado Anschutz Medical Campus, Aurora, CO, United States of America, 7 Department of Statistics, Oklahoma State University, Stillwater, OK, United States of America * [email protected] OPEN ACCESS Citation: Radcliffe RA, Dowell R, Odell AT, Richmond PA, Bennett B, Larson C, et al. (2020) Abstract Systems genetics analysis of the LXS recombinant inbred mouse strains:Genetic and molecular We have been using the Inbred Long- and Short-Sleep mouse strains (ILS, ISS) and a insights into acute ethanol tolerance. PLoS ONE recombinant inbred panel derived from them, the LXS, to investigate the genetic underpin- 15(10): e0240253. https://doi.org/10.1371/journal. -

SDHAF1 Antibody (Center) Affinity Purified Rabbit Polyclonal Antibody (Pab) Catalog # AP5392C
10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 SDHAF1 Antibody (Center) Affinity Purified Rabbit Polyclonal Antibody (Pab) Catalog # AP5392C Specification SDHAF1 Antibody (Center) - Product Information Application WB, IHC-P,E Primary Accession A6NFY7 Other Accession B0K036, Q3U276, A8PU71, NP_001036096.1 Reactivity Human Predicted Bovine, Mouse, Rat Host Rabbit Clonality Polyclonal Isotype Rabbit Ig Calculated MW 12806 Antigen Region 34-62 SDHAF1 Antibody (Center)(Cat. #AP5392c) SDHAF1 Antibody (Center) - Additional western blot analysis in ZR-75-1 cell line Information lysates (35ug/lane).This demonstrates the SDHAF1 antibody detected the SDHAF1 Gene ID 644096 protein (arrow). Other Names Succinate dehydrogenase assembly factor 1, mitochondrial, SDH assembly factor 1, SDHAF1, LYR motif-containing protein 8, SDHF1 Target/Specificity This SDHAF1 antibody is generated from rabbits immunized with a KLH conjugated synthetic peptide between 34-62 amino acids from the Central region of human SDHAF1. Dilution SDHAF1 Antibody (Center) (Cat. #AP5392c) WB~~1:1000 immunohistochemistry analysis in formalin IHC-P~~1:50~100 fixed and paraffin embedded human prostate carcinoma followed by peroxidase Format conjugation of the secondary antibody and Purified polyclonal antibody supplied in PBS DAB staining. This data demonstrates the use with 0.09% (W/V) sodium azide. This of the SDHAF1 Antibody (Center) for antibody is purified through a protein A immunohistochemistry. Clinical relevance has column, followed by peptide affinity not been evaluated. purification. Storage SDHAF1 Antibody (Center) - Background Maintain refrigerated at 2-8°C for up to 2 Page 1/3 10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 weeks. -

Gnomad Lof Supplement
1 gnomAD supplement gnomAD supplement 1 Data processing 4 Alignment and read processing 4 Variant Calling 4 Coverage information 5 Data processing 5 Sample QC 7 Hard filters 7 Supplementary Table 1 | Sample counts before and after hard and release filters 8 Supplementary Table 2 | Counts by data type and hard filter 9 Platform imputation for exomes 9 Supplementary Table 3 | Exome platform assignments 10 Supplementary Table 4 | Confusion matrix for exome samples with Known platform labels 11 Relatedness filters 11 Supplementary Table 5 | Pair counts by degree of relatedness 12 Supplementary Table 6 | Sample counts by relatedness status 13 Population and subpopulation inference 13 Supplementary Figure 1 | Continental ancestry principal components. 14 Supplementary Table 7 | Population and subpopulation counts 16 Population- and platform-specific filters 16 Supplementary Table 8 | Summary of outliers per population and platform grouping 17 Finalizing samples in the gnomAD v2.1 release 18 Supplementary Table 9 | Sample counts by filtering stage 18 Supplementary Table 10 | Sample counts for genomes and exomes in gnomAD subsets 19 Variant QC 20 Hard filters 20 Random Forest model 20 Features 21 Supplementary Table 11 | Features used in final random forest model 21 Training 22 Supplementary Table 12 | Random forest training examples 22 Evaluation and threshold selection 22 Final variant counts 24 Supplementary Table 13 | Variant counts by filtering status 25 Comparison of whole-exome and whole-genome coverage in coding regions 25 Variant annotation 30 Frequency and context annotation 30 2 Functional annotation 31 Supplementary Table 14 | Variants observed by category in 125,748 exomes 32 Supplementary Figure 5 | Percent observed by methylation. -
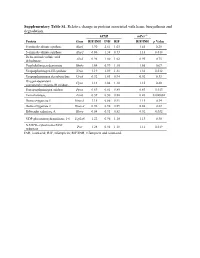
Supplementary Table S1. Relative Change in Proteins Associated with Heme Biosynthesis and Degradation
Supplementary Table S1. Relative change in proteins associated with heme biosynthesis and degradation. hPXR mPxr–/– Protein Gene RIF/INH INH RIF RIF/INH p Value 5-aminolevulinate synthase Alas1 1.90 2.61 1.05 1.41 0.28 5-aminolevulinate synthase Alas2 0.86 1.38 0.73 1.18 0.018 Delta-aminolevulinic acid Alad 0.96 1.00 1.02 0.95 0.75 dehydratase Porphobilinogen deaminase Hmbs 1.04 0.99 1.10 1.05 0.67 Uroporphyrinogen-III synthase Uros 1.19 1.09 1.31 1.38 0.012 Uroporphyrinogen decarboxylase Urod 0.92 1.03 0.94 0.92 0.33 Oxygen-dependent Cpox 1.13 1.04 1.18 1.15 0.20 coproporphyrinogen-III oxidase, Protoporphyrinogen oxidase Ppox 0.69 0.81 0.85 0.83 0.013 Ferrochelatase, Fech 0.39 0.50 0.88 0.43 0.000002 Heme oxygenase 1 Hmox1 1.15 0.86 0.91 1.11 0.34 Heme oxygenase 2 Hmox2 0.96 0.98 0.89 0.88 0.22 Biliverdin reductase A Blvra 0.84 0.92 0.82 0.92 0.032 UDP-glucuronosyltransferase 1-6 Ugt1a6 1.22 0.96 1.10 1.13 0.30 NADPH--cytochrome P450 Por 1.28 0.92 1.18 1.12 0.019 reductase INH, isoniazid; RIF, rifampicin; RIF/INH, rifampicin and isoniazid. Supplementary Table S2. Relative change in protein nuclear receptors. hPXR mPxr–/– Protein Gene RIF/INH INH RIF RIF/INH p Value Aryl hydrocarbon receptor Ahr 1.09 0.91 1.00 1.26 0.092 Hepatocyte nuclear factor Hnf1a 0.87 0.97 0.82 0.79 0.027 1-alpha Hepatocyte nuclear factor Hnf4a 0.95 1.05 0.97 1.08 0.20 4-alpha Oxysterols receptor LXR- Nr1h3 0.94 1.16 1.03 1.02 0.42 alpha Bile acid receptor Nr1h4 1.05 1.17 0.98 1.19 0.12 Retinoic acid receptor Rxra 0.88 1.03 0.83 0.95 0.12 RXR-alpha Peroxisome proliferator- -

The Role of Immunohistochemistry and Molecular Analysis of Succinate Dehydrogenase in the Diagnosis of Endocrine and Non-Endocrine Tumors and Related Syndromes
Endocrine Pathology https://doi.org/10.1007/s12022-018-9555-2 The Role of Immunohistochemistry and Molecular Analysis of Succinate Dehydrogenase in the Diagnosis of Endocrine and Non-Endocrine Tumors and Related Syndromes Lindsey Oudijk1 & José Gaal2 & Ronald R. de Krijger3 # The Author(s) 2018 Abstract Succinate dehydrogenase (SDH) is an enzyme complex, composed of four protein subunits, that plays a role in both the citric acid cycle and the electron transport chain. The genes for SDHA, SDHB, SDHC, and SDHD are located in the nuclear DNA, and mutations in these genes have initially been described in paragangliomas (PGL) and pheochromocytomas (PCC), which are relatively rare tumors derived from the autonomic nervous system and the adrenal medulla, respectively. Patients with SDH mutations, that are almost exclusively in the germline, are frequently affected by multiple PGL and/or PCC. In addition, other tumors have been associated with SDH mutations as well, including gastrointestinal stromal tumors, SDH-deficient renal cell carcinoma, and pituitary adenomas. Immunohistochemistry for SDHB and SDHA has been shown to be a valuable additional tool in the histopathological analysis of these tumors, and can be considered as a surrogate marker for molecular analysis. In addition, SDHB immunohistochemistry is relevant in the decision-making whether a genetic sequence variant represents a pathogenic mutation or not. In this review, we highlight the current knowledge of the physiologic and pathologic role of the SDH enzyme complex and its involvement in endocrine and non-endocrine tumors, with an emphasis on the applicability of immunohistochemistry. Keywords Pheochromocytoma . Paraganglioma . Gastrointestinal stromal tumor . Histology . Genetics . Succinate dehydrogenase SDH Complex: Genes, Proteins, Function and the binding site for succinate, and SDH subunit B (SDHB), an iron-sulfur protein with three FeS clusters. -

SDHAF1 Antibody Cat
SDHAF1 Antibody Cat. No.: 6879 SDHAF1 Antibody Immunocytochemistry of SDHAF1 in 3T3 cells with Immunofluorescence of SDHAF1 in 3T3 cells with SDHAF1 SDHAF1 antibody at 5 μg/mL. antibody at 20 μg/mL. Specifications HOST SPECIES: Rabbit SPECIES REACTIVITY: Human, Mouse SDHAF1 antibody was raised against an 18 amino acid synthetic peptide near the carboxy terminus of human SDHAF1. IMMUNOGEN: The immunogen is located within the last 50 amino acids of SDHAF1. TESTED APPLICATIONS: ELISA, ICC, IF, WB October 2, 2021 1 https://www.prosci-inc.com/sdhaf1-antibody-6879.html SDHAF1 antibody can be used for detection of SDHAF1 by Western blot at 1 - 2 μg/mL. Antibody can also be used for immunocytochemistry starting at 5 μg/mL. For immunofluorescence start at 5 μg/mL. APPLICATIONS: Antibody validated: Western Blot in mouse samples; Immunocytochemistry in mouse samples and Immunofluorescence in mouse samples. All other applications and species not yet tested. SDHAF1 antibody is predicted to not cross-react with other SDHAF protein family SPECIFICITY: members. POSITIVE CONTROL: 1) Cat. No. 1282 - 3T3 (NIH) Cell Lysate 2) Cat. No. 17-201 - 3T3/BALB Cell Slide Properties PURIFICATION: SDHAF1 Antibody is affinity chromatography purified via peptide column. CLONALITY: Polyclonal ISOTYPE: IgG CONJUGATE: Unconjugated PHYSICAL STATE: Liquid BUFFER: SDHAF1 Antibody is supplied in PBS containing 0.02% sodium azide. CONCENTRATION: 1 mg/mL SDHAF1 antibody can be stored at 4˚C for three months and -20˚C, stable for up to one STORAGE CONDITIONS: year. As with all antibodies care should be taken to avoid repeated freeze thaw cycles. Antibodies should not be exposed to prolonged high temperatures.