Karyotype Aberrations in Action: the Evolution of Cancer Genomes and the Tumor Microenvironment
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Timing of Centrosome Separation Is Important for Accurate Chromosome Segregation
M BoC | ARTICLE Timing of centrosome separation is important for accurate chromosome segregation William T. Silkwortha, Isaac K. Nardia,*, Raja Paulb, Alex Mogilnerc, and Daniela Ciminia aDepartment of Biological Sciences, Virginia Tech, Blacksburg, VA 24061; bIndian Association for the Cultivation of Science, Jadavpur, Kolkata 700032, India; cDepartment of Neurobiology, Physiology and Behavior and Department of Mathematics, University of California, Davis, Davis, CA 95616 ABSTRACT Spindle assembly, establishment of kinetochore attachment, and sister chroma- Monitoring Editor tid separation must occur during mitosis in a highly coordinated fashion to ensure accurate Yixian Zheng chromosome segregation. In most vertebrate cells, the nuclear envelope must break down to Carnegie Institution allow interaction between microtubules of the mitotic spindle and the kinetochores. It was Received: Feb 2, 2011 previously shown that nuclear envelope breakdown (NEB) is not coordinated with centrosome Revised: Nov 17, 2011 separation and that centrosome separation can be either complete at the time of NEB or can Accepted: Nov 22, 2011 be completed after NEB. In this study, we investigated whether the timing of centrosome separation affects subsequent mitotic events such as establishment of kinetochore attach- ment or chromosome segregation. We used a combination of experimental and computa- tional approaches to investigate kinetochore attachment and chromosome segregation in cells with complete versus incomplete spindle pole separation at NEB. We found that cells with incomplete spindle pole separation exhibit higher rates of kinetochore misattachments and chromosome missegregation than cells that complete centrosome separation before NEB. Moreover, our mathematical model showed that two spindle poles in close proximity do not “search” the entire cellular space, leading to formation of large numbers of syntelic at- tachments, which can be an intermediate stage in the formation of merotelic kinetochores. -

SETD2 Haploinsufficiency for Microtubule Methylation Is an Early Driver of Genomic Instability in Renal Cell Carcinoma
Published OnlineFirst May 3, 2018; DOI: 10.1158/0008-5472.CAN-17-3460 Cancer Genome and Epigenome Research SETD2 Haploinsufficiency for Microtubule Methylation Is an Early Driver of Genomic Instability in Renal Cell Carcinoma Yun-Chen Chiang1, In-Young Park2, Esteban A. Terzo3, Durga Nand Tripathi2, Frank M. Mason3, Catherine C. Fahey1, Menuka Karki2,4, Charles B. Shuster4, Bo-Hwa Sohn2, Pratim Chowdhury2, Reid T. Powell5, Ryoma Ohi6, Yihsuan S. Tsai1, Aguirre A. de Cubas3, Abid Khan1,7, Ian J. Davis1, Brian D. Strahl1,7, Joel S. Parker1, Ruhee Dere2, Cheryl L. Walker2, and W. Kimryn Rathmell3 Abstract Loss of the short arm of chromosome 3 (3p) occurs early in human kidney cells, rescue with a pathogenic SETD2 mutant >95% of clear cell renal cell carcinoma (ccRCC). Nearly ubiqui- deficient for microtubule (aTubK40me3), but not histone tous 3p loss in ccRCC suggests haploinsufficiency for 3p tumor (H3K36me3) methylation, replicated this phenotype. Genomic suppressors as early drivers of tumorigenesis. We previously instability (micronuclei) was also a hallmark of patient-derived reported methyltransferase SETD2, which trimethylates H3 his- cells from ccRCC. These data show that the SETD2 tumor sup- tones on lysine 36 (H3K36me3) and is located in the 3p deletion, pressor displays a haploinsufficiency phenotype disproportion- to also trimethylate microtubules on lysine 40 (aTubK40me3) ately impacting microtubule methylation and serves as an early during mitosis, with aTubK40me3 required for genomic sta- driver of genomic instability. bility. We now show that monoallelic, Setd2-deficient cells retain- Significance: Loss of a single allele of a chromatin modifier ing H3K36me3, but not aTubK40me3, exhibit a dramatic plays a role in promoting oncogenesis, underscoring the grow- increase in mitotic defects and micronuclei count, with increased ing relevance of tumor suppressor haploinsufficiency in tumor- viability compared with biallelic loss. -

GENETICS and GENOMICS Ed
GENETICS AND GENOMICS Ed. Csaba Szalai, PhD GENETICS AND GENOMICS Editor: Csaba Szalai, PhD, university professor Authors: Chapter 1: Valéria László Chapter 2, 3, 4, 6, 7: Sára Tóth Chapter 5: Erna Pap Chapter 8, 9, 10, 11, 12, 13, 14: Csaba Szalai Chapter 15: András Falus and Ferenc Oberfrank Keywords: Mitosis, meiosis, mutations, cytogenetics, epigenetics, Mendelian inheritance, genetics of sex, developmental genetics, stem cell biology, oncogenetics, immunogenetics, human genomics, genomics of complex diseases, genomic methods, population genetics, evolution genetics, pharmacogenomics, nutrigenetics, gene environmental interaction, systems biology, bioethics. Summary The book contains the substance of the lectures and partly of the practices of the subject of ‘Genetics and Genomics’ held in Semmelweis University for medical, pharmacological and dental students. The book does not contain basic genetics and molecular biology, but rather topics from human genetics mainly from medical point of views. Some of the 15 chapters deal with medical genetics, but the chapters also introduce to the basic knowledge of cell division, cytogenetics, epigenetics, developmental genetics, stem cell biology, oncogenetics, immunogenetics, population genetics, evolution genetics, nutrigenetics, and to a relative new subject, the human genomics and its applications for the study of the genomic background of complex diseases, pharmacogenomics and for the investigation of the genome environmental interactions. As genomics belongs to sytems biology, a chapter introduces to basic terms of systems biology, and concentrating on diseases, some examples of the application and utilization of this scientific field are also be shown. The modern human genetics can also be associated with several ethical, social and legal issues. The last chapter of this book deals with these issues. -

Pedigrees and Karyotypes Pedigree
Pedigrees and Karyotypes Pedigree A pedigree shows the relationships within a family and it helps to chart how one gene can be passed on from generation to generation. Pedigrees are tools used by genetic researchers or counselors to identify a genetic condition running through a family, they aid in making a diagnosis, and aid in determining who in the family is at risk for genetic conditions. On a pedigree: A circle represents a female A square represents a male A horizontal line connecting a male and female represents a marriage A vertical line and a bracket connect the parents to their children A circle/square that is shaded means the person HAS the trait. A circle/square that is not shaded means the person does not have the trait. Children are placed from oldest to youngest. A key is given to explain what the trait is. Marriage Male-DAD Female-MOM Has the trait Male-Son Female-daughter Female-daughter Male- Son Oldest to youngest Steps: ff Ff •Identify all people who have the trait. •For the purpose of this class all traits will be given to you. In other instances, you would have to determine whether or not the trait is autosomal dominant, autosomal recessive, or sex- linked. •In this example, all those who have the trait are homozygous recessive. •Can you correctly identify all genotypes of this family? ff ff Ff Ff •F- Normal •f- cystic fibrosis Key: affected male affected female unaffected male unaffected female Pp Pp PKU P- Unaffected p- phenylketonuria PP or Pp pp Pp pp pp Pp Pp Key: affected male affected female unaffected male unaffected female H-huntington’s hh Hh disease h-Unaffected Hh hh Hh hh Hh hh hh Key: affected male affected female unaffected male unaffected female Sex-Linked Inheritance Colorblindness Cy cc cy Cc Cc cy cy Key: affected male affected female unaffected male unaffected female Karyotypes To analyze chromosomes, cell biologists photograph cells in mitosis, when the chromosomes are fully condensed and easy to see (usually in metaphase). -
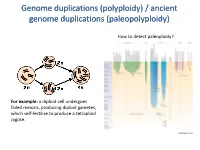
Polyploidy) / Ancient Genome Duplications (Paleopolyploidy
Genome duplications (polyploidy) / ancient genome duplications (paleopolyploidy) How to detect paleoploidy? For example: a diploid cell undergoes failed meiosis, producing diploid gametes, which self-fertilize to produce a tetraploid zygote. Timing of duplication by trees (phylogenetic timing) Phylogenetic timing of duplicates b Paramecium genome duplications Comparison of two scaffolds originating from a common ancestor at the recent WGD Saccharomyces cerevisiae Just before genome duplication Just after genome duplication More time after genome duplication Unaligned view (removing gaps just like in cerev has occurred) Saccharomyces cerevisiae Problem reciprocal gene loss (extreme case); how to solve? Problem reciprocal gene loss (extreme case); how to solve? Just before genome duplication Outgroup! Just after genome duplication Outgroup Just after genome duplication Outgroup More time after genome duplication Outgroup Problem (extreme case); how to solve? Outgroup Outgroup Outgroup Outgroup Outgroup Using other genomes Wong et al. 2002 PNAS Centromeres Vertebrate genome duplication Nature. 2011 Apr 10. [Epub ahead of print] Ancestral polyploidy in seed plants and angiosperms. Jiao Y, Wickett NJ, Ayyampalayam S, Chanderbali AS, Landherr L, Ralph PE, Tomsho LP, Hu Y, Liang H, Soltis PS, Soltis DE, Clifton SW, Schlarbaum SE, Schuster SC, Ma H, Leebens-Mack J, Depamphilis CW. Flowering plants Flowering MOSS Vertebrates Teleosts S. serevisiae and close relatives Paramecium Reconstructed map of genome duplications allows unprecedented mapping -

The Diversity of Plant Sex Chromosomes Highlighted Through Advances in Genome Sequencing
G C A T T A C G G C A T genes Review The Diversity of Plant Sex Chromosomes Highlighted through Advances in Genome Sequencing Sarah Carey 1,2 , Qingyi Yu 3,* and Alex Harkess 1,2,* 1 Department of Crop, Soil, and Environmental Sciences, Auburn University, Auburn, AL 36849, USA; [email protected] 2 HudsonAlpha Institute for Biotechnology, Huntsville, AL 35806, USA 3 Texas A&M AgriLife Research, Texas A&M University System, Dallas, TX 75252, USA * Correspondence: [email protected] (Q.Y.); [email protected] (A.H.) Abstract: For centuries, scientists have been intrigued by the origin of dioecy in plants, characterizing sex-specific development, uncovering cytological differences between the sexes, and developing theoretical models. Through the invention and continued improvements in genomic technologies, we have truly begun to unlock the genetic basis of dioecy in many species. Here we broadly review the advances in research on dioecy and sex chromosomes. We start by first discussing the early works that built the foundation for current studies and the advances in genome sequencing that have facilitated more-recent findings. We next discuss the analyses of sex chromosomes and sex-determination genes uncovered by genome sequencing. We synthesize these results to find some patterns are emerging, such as the role of duplications, the involvement of hormones in sex-determination, and support for the two-locus model for the origin of dioecy. Though across systems, there are also many novel insights into how sex chromosomes evolve, including different sex-determining genes and routes to suppressed recombination. We propose the future of research in plant sex chromosomes should involve interdisciplinary approaches, combining cutting-edge technologies with the classics Citation: Carey, S.; Yu, Q.; to unravel the patterns that can be found across the hundreds of independent origins. -

Possible Hazards of Cell Phones and Towers, Wi-Fi, Smart Meters, and Wireless Computers, Printers, Laptops, Mice, Keyboards, and Routers Book Four
Possible Hazards of Cell Phones and Towers, Wi-Fi, Smart Meters, and Wireless Computers, Printers, Laptops, Mice, Keyboards, and Routers Book Four Since 2013 I have been emailed several dozen reports of possible medical and other hazards from intense electromagnetic radiation from cell phones and towers, Wi-Fi, smart meters, and wireless computer accessories including wireless computers, keyboards, mice, routers, printers, and laptops. I have previously compiled a total of 600 pages of these reports in chronological order in three separate books with the same title as this “Book Four”. All four ‘EMF Hazards’ books are linked at www.commutefaster.com/vesperman.html and www.padrak.com/vesperman. Approximately 35 authoritative wireless radiation hazards-related reports are also linked at these two websites. This report begins with “Disclaimers”, a table of contents, “Items of Outstanding Interest”, and a new supplementary set of potentially useful “Recommendations for Actions”. Gary Vesperman 588 Lake Huron Lane Boulder City, NV 89005-1018 702-435-7947 [email protected] Hazards of Cell Phones, Wireless Devices, Etc – Book Four 1 December 14, 2016 Disclaimers Inclusion of any invention or technology in this “Possible Hazards of Cell Phones and Towers, Wi-Fi, Smart Meters, and Wireless Computers, Printers, Laptops, Mice, Keyboards, and Routers – Book Four” does not in any way imply its suitability for investments of any kind. Nor does inclusion of any invention or technology described or mentioned herein conclusively implies safety or hazards. Gary C. Vesperman, Boulder City, Nevada and the numerous contributors to this compilation do not warrant that any of the information presented is accurate, complete, and not misleading. -

Double Mitotic Nondisjunction
J Med Genet: first published as 10.1136/jmg.15.5.395 on 1 October 1978. Downloaded from Case reports 395 well be that, in this family, 18q- is unable to segre- mosaicism. Chromosome 18 trisomy was found gate properly during gametogenesis and early post- only in 18% of lymphocytes and not in skin zygotic mitosis, leading to an unbalanced state and fibroblasts. A likely interpretation is double non- +(18q-). disjunction in a single lymphocyte precursor HAROLD N. BASs, FELICE WEBER-PARISI, AND of a trisomy 21 embryo. A brief review of other ROBERT S. SPARKES cases of mitotic multiple nondisjunction and Department ofPediatrics (Genetics), Kaiser- double aneuploid mosaicism is presented. Permanente Medical Center, Panorama City, California 91402; and Division ofMedical Genetics, Chromosomal mosaicism occurs in a small percentage Departments ofMedicine, Pediatrics, andPsychiatry, of patients with Down's syndrome. The vast majority UCLA Centerfor the Health Sciences, Los Angeles, of these mosaics have a normal cell line in addition to California 90024, USA the trisomy 21 cell line. This report describes the first reported instance, to our knowledge, of an unusual References type of double trisomy mosaicism in lymphocytes Castel, Y., Riviere, D., Nawrocki, T., Le Fur, J-M., and Toudic, involving chromosomes 18 and 21. L. (1975). Trisomie partielie 18q par translocation familiale t(l8q-;13q+). Lyon Medical, 233, 211-217. Francke, U. (1972). Quinacrine mustard fluorescence of human chromosomes: characterization of unusual translocations. Case report American Journal ofHuman Genetics, 24, 189-213. Fujita, K., and Fujita, H. M. (1974). An extra small submetacentric The proposita was referred for chromosome analysis chromosome: possible partial trisomy 18. -

Epigenetic Centromere Specification Directs Aurora B Accumulation but Is Insufficient to Efficiently Correct Mitotic Errors
JCB: Report Epigenetic centromere specification directs aurora B accumulation but is insufficient to efficiently correct mitotic errors Emily A. Bassett,1,2 Stacey Wood,2 Kevan J. Salimian,1,2 Sandya Ajith,1,2 Daniel R. Foltz,3 and Ben E. Black1,2 1Graduate Group in Biochemistry and Molecular Biophysics and 2Department of Biochemistry and Biophysics, School of Medicine, University of Pennsylvania, Philadelphia, PA 19104 3Department of Biochemistry and Molecular Genetics, University of Virginia, Charlottesville, VA 22908 he nearly ubiquitous presence of repetitive centro- of the kinetochore is intact at a human neocentromere mere DNA sequences across eukaryotic species is in lacking repetitive -satellite DNA. However, aurora B is Tparadoxical contrast to their apparent functional dis- inappropriately silenced as a consequence of the altered pensability. Centromeric chromatin is spatially delineated geometry of the neocentromere, thereby compromising into the kinetochore-forming array of centromere protein A the error correction mechanism. This suggests a model (CENP-A)–containing nucleosomes and the inner cen wherein the neocentromere represents a primordial inher- tromeric heterochromatin that lacks CENP-A but recruits itance locus that requires subsequent generation of a the aurora B kinase that is necessary for correcting erro- robust inner centromere compartment to enhance fidelity neous attachments to the mitotic spindle. We found that of chromosome transmission. the self-perpetuating network of CENPs at the foundation Introduction -

Chromosome Missegregation in Human Cells Arises Through Specific
Chromosome missegregation in human cells arises through specific types of kinetochore–microtubule attachment errors Sarah L. Thompson and Duane A. Compton1 Department of Biochemistry, Dartmouth Medical School, Hanover, NH 03755; and Cancer Mechanisms Program, Norris Cotton Cancer Center, Lebanon, NH 03766 Edited by John W. Sedat, University of California, San Francisco School of Medicine, San Francisco, CA, and approved September 15, 2011 (received for review June 17, 2011) Most solid tumors are aneuploid, and many missegregate chro- cur, particularly in early phases of mitosis, as a consequence mosomes at high rates in a phenomenon called chromosomal of the stochastic interactions between microtubules and kinet- instability (CIN). CIN reflects the erosion of mitotic fidelity, and it ochores (11). A prominent error is when a single kinetochore correlates with poor patient prognosis and drug resistance. The binds microtubules oriented toward both spindle poles. This most common mechanism causing CIN is the persistence of improper error is called merotely (12, 13). The persistence of merotely kinetochore–microtubule attachments called merotely. Chromo- undermines chromosome segregation because merotelic kinet- somes with merotelic kinetochores often manifest as lagging chro- ochores experience poleward force toward both spindle poles. As mosomes in anaphase, suggesting that lagging chromosomes fail a consequence, merotely often results in the appearance of lag- to segregate properly. However, it remains unknown whether the ging chromosomes in anaphase, and tumor cells with CIN have lagging chromosomes observed in anaphase segregate to the cor- elevated rates of lagging chromosomes and merotelic attach- rect or incorrect daughter cell. To address this question, we tracked ments (9). Moreover, it was shown that increasing the correction the segregation of a single human chromosome during cell division rate of merotely by stimulating the dynamics of k-MT attachment by using LacI-GFP to target an integrated LacO array. -

Cytogenetics, Chromosomal Genetics
Cytogenetics Chromosomal Genetics Sophie Dahoun Service de Génétique Médicale, HUG Geneva, Switzerland [email protected] Training Course in Sexual and Reproductive Health Research Geneva 2010 Cytogenetics is the branch of genetics that correlates the structure, number, and behaviour of chromosomes with heredity and diseases Conventional cytogenetics Molecular cytogenetics Molecular Biology I. Karyotype Definition Chromosomal Banding Resolution limits Nomenclature The metaphasic chromosome telomeres p arm q arm G-banded Human Karyotype Tjio & Levan 1956 Karyotype: The characterization of the chromosomal complement of an individual's cell, including number, form, and size of the chromosomes. A photomicrograph of chromosomes arranged according to a standard classification. A chromosome banding pattern is comprised of alternating light and dark stripes, or bands, that appear along its length after being stained with a dye. A unique banding pattern is used to identify each chromosome Chromosome banding techniques and staining Giemsa has become the most commonly used stain in cytogenetic analysis. Most G-banding techniques require pretreating the chromosomes with a proteolytic enzyme such as trypsin. G- banding preferentially stains the regions of DNA that are rich in adenine and thymine. R-banding involves pretreating cells with a hot salt solution that denatures DNA that is rich in adenine and thymine. The chromosomes are then stained with Giemsa. C-banding stains areas of heterochromatin, which are tightly packed and contain -

Mitosis Meiosis Karyotype
POGIL Cell Biology Activity 7 – Meiosis/Gametogenesis Schivell MODEL 1: karyotype Meiosis Mitosis 1 POGIL Cell Biology Activity 7 – Meiosis/Gametogenesis Schivell MODEL 2, Part 1: Spermatogenesis The trapezoid below represents a small portion of the wall of a "seminiferous tubule" within the testis. The cells in each of the panels are all originally derived from the single cell in panel 1. 1 2 3 Outside of tubule Lumen of tubule 4 5 6 7 8 9 2 POGIL Cell Biology Activity 7 – Meiosis/Gametogenesis Schivell MODEL 2, Part 2: vas epididymis deferens testis (plural: testes) seminiferous tubules (cut) Courtesy of: Dr. E. Kent Christensen, U. of Michigan lumen of seminiferous tubule sperm This portion shown expanded in part 1 of Model 2 3 POGIL Cell Biology Activity 7 – Meiosis/Gametogenesis Schivell MODEL 3: Oogenesis This is a time lapse of an ovary showing one "follicle" as it develops from immaturity to ovulation. The follicle starts in panel 1 as a small sphere of "follicle cells" surrounding the oocyte. In each panel, chromosomes within the oocyte are shown as an inset. (There are actually thousands of follicles in each mammalian ovary). 1 2 3 4 5 6 7 4 POGIL Cell Biology Activity 7 – Meiosis/Gametogenesis Schivell Model 1 questions: 1. Using the same type of cartoon as model 1, draw an "unreplicated", condensed chromosome. 2. Draw a replicated, condensed chromosome: 3. Circle a homologous pair in the karyotype. Remember that one of these chromosomes came from the male parent and the other from the female parent. These two chromosomes carry the same genes! (But can have different alleles on each homolog.) 4.