Drying-Induced Atomic Structural Rearrangements in Sodium-Based
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

White Concrete & Colored Concrete Construction
WHITE CEMENT CONCRETE AND COLORED CONCRETE CONSTRUCTION and developers Owners seeking dynamic architectural results demand the benefits that only white or colored concrete can deliver. Informed designers rely on careful selection of materials and attention to mixing and placement details to achieve eye-catching architectural and decorative finishes. Because of its versatility, concrete offers endless design possibilities. A wide variety of decorative looks are achieved by using colored aggregates and pigments, and by varying surface finishes, treatments, and textures. Combining white cement with pigments and colored aggregates expands the number of colors available to discerning customers. Colors are cleaner and more intense because pigments and specialty Figure 1. The Condell Medical Center is built with load-bearing precast concrete aggregates don’t have to overcome the panels with an architectural finish achieved by using white cement. grayish paste of common concrete. Courtesy of Condell Medical Center. Mix Designs • CSA A3000 cements that conform to the Canadian From a structural design perspective, white cement-based Standards Association standards for Cementitious Materials concrete mixes are identical to gray cement-based mixes. Mix • American Association of State Highway and Transportation designs for white or colored concrete differ from common Officials, AASHTO M85 Standard Specification for Portland concrete in that they are formulated based on each ingredient’s Cement effect on concrete color. Listed in order of decreasing -
Cement Data Sheet
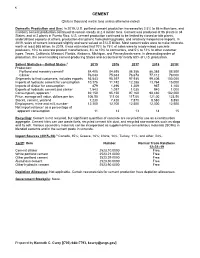
42 CEMENT (Data in thousand metric tons unless otherwise noted) Domestic Production and Use: In 2019, U.S. portland cement production increased by 2.5% to 86 million tons, and masonry cement production continued to remain steady at 2.4 million tons. Cement was produced at 96 plants in 34 States, and at 2 plants in Puerto Rico. U.S. cement production continued to be limited by closed or idle plants, underutilized capacity at others, production disruptions from plant upgrades, and relatively inexpensive imports. In 2019, sales of cement increased slightly and were valued at $12.5 billion. Most cement sales were to make concrete, worth at least $65 billion. In 2019, it was estimated that 70% to 75% of sales were to ready-mixed concrete producers, 10% to concrete product manufactures, 8% to 10% to contractors, and 5% to 12% to other customer types. Texas, California, Missouri, Florida, Alabama, Michigan, and Pennsylvania were, in descending order of production, the seven leading cement-producing States and accounted for nearly 60% of U.S. production. Salient Statistics—United States:1 2015 2016 2017 2018 2019e Production: Portland and masonry cement2 84,405 84,695 86,356 86,368 88,500 Clinker 76,043 75,633 76,678 77,112 78,000 Shipments to final customers, includes exports 93,543 95,397 97,935 99,406 100,000 Imports of hydraulic cement for consumption 10,376 11,742 12,288 13,764 15,000 Imports of clinker for consumption 879 1,496 1,209 967 1,100 Exports of hydraulic cement and clinker 1,543 1,097 1,035 940 1,000 Consumption, apparent3 92,150 95,150 97,160 98,480 102,000 Price, average mill value, dollars per ton 106.50 111.00 117.00 121.00 123.50 Stocks, cement, yearend 7,230 7,420 7,870 8,580 8,850 Employment, mine and mill, numbere 12,300 12,700 12,500 12,300 12,500 Net import reliance4 as a percentage of apparent consumption 11 13 13 14 15 Recycling: Cement is not recycled, but significant quantities of concrete are recycled for use as a construction aggregate. -

Gray Portland Cement and Cement Clinker from Japan
Gray Portland Cement and Cement Clinker from Japan Investigation No. 731-TA-461 (Fourth Review) Publication 4704 June 2017 U.S. International Trade Commission Washington, DC 20436 U.S. International Trade Commission COMMISSIONERS Rhonda K. Schmidtlein, Chairman David S. Johanson, Vice Chairman Irving A. Williamson Meredith M. Broadbent F. Scott Kieff Catherine DeFilippo Director of Operations Staff assigned Carolyn Carlson, Investigator Gregory LaRocca, Industry Analyst Andrew Knipe, Economist Heng Loke, Attorney Fred Ruggles, Supervisory Investigator Address all communications to Secretary to the Commission United States International Trade Commission Washington, DC 20436 U.S. International Trade Commission Washington, DC 20436 www.usitc.gov Gray Portland Cement and Cement Clinker from Japan Investigation No. 731-TA-461 (Fourth Review) Publication 4704 June 2017 CONTENTS Page Determination .............................................................................................................................1 Views of the Commission .............................................................................................................3 Information obtained in the review ........................................................................................... I-1 Background ...................................................................................................................................... I-1 Responses to the Commission's notice of institution ..................................................................... -

Portland Cements CAS Reg
3251 Bath Pike Nazareth, PA 18064 MATERIAL SAFETY DATA SHEET Section 1 - IDENTIFICATION Product Name: Portland Cements CAS Reg. No.: 65997-15-1 Chemical Name and Synonyms: Portland Cement, Cement, Hydraulic Cement Trade Names: Portland Cement – Types I, IA, II, III, IIIA, GU, MS,LH, HE; SAYLOR’S Portland Types: I, IA, II, III; PRONTO, Flamingo Brixment White Portland Cement; Oil Well Cement Class A, H. MSDS Information: This MSDS supersedes prior MSDS’s for the products noted above. This MSDS covers a number of products with similar applications and occupational exposure hazards. Specific constituents and methods of preparation for these products will vary. The term “Portland Cement”, used in the text of this MSDS, refers to the above named products collectively. Chemical Family: Calcium silicate compounds; calcium compounds containing iron and aluminum; and gypsum are the primary constituents of these products. Informational Phone Numbers: (800) 437-7762 Customer Service - Nazareth, PA (800) 336-0366 Customer Service - Speed, IN (800) 624-8986 Customer Service - Martinsburg, WV (800) 386-2111 Customer Service - Mississauga, ONT Emergency Contact Information: (800)-424-9300 Chemtrec MSDS Prepared by: Essroc MSDS Development Committee - (610) 837-6725 – May 9, 2013 Section 2 - COMPONENTS Hazardous Ingredients: Component CAS No. OSHA PEL (8-hour TWA) ACGIH TLV Other Information Portland Cement 65997-15-1 15 mg total dust/m3 1.0 mg/m3 IDLH: 5000 mg/m3 3 5 mg respirable dust/m respirable LD50: No Data Gypsum 13397-24-5 15 mg total dust/m3 -

WHITE PORTLAND CEMENT TYPE I Durable, Dependable, Cost-Effective Basic Building Material
WHITE PORTLAND CEMENT TYPE I Durable, dependable, cost-effective basic building material 1. PRODUCT NAME Mixing Tech-Mix® White Portland Cement (Type I) Refer to the mix designs chart for typical proportions by volume per appropriate application. Add the minimum amount 2. MANUFACTURER of clean water necessary, too much water will weaken the Tech-Mix® is a registered trademark of TCC Materials product. Mix until uniform, workable consistency is reached. 2025 Centre Pointe Blvd., Suite 300 Mendota Heights, MN 55120 USA Typical Mix Designs by Volume Phone: 1.651.688.9116 Web: techmixpro.com Concrete Mix Portland Cement General Purpose Gravel or Stone 3. PRODUCT DESCRIPTION Sand (ASTM C-33) (ASTM C-33) Tech-Mix ® White Portland Cement Type I are regularly used to produce architectural concrete. General applications include 1 Part 2 Parts 3 Parts cast-in-place or precast wall panels, floors, slabs, terrazzo, thin Mortar Mix (Type S, ASTM C-270) -set and tile grouts, cast stone, masonry units and mortars, stucco, swimming pools & spas, glass fiber reinforced Portland Cement Hydrated Lime Masonry Sand concrete, ornamental statuary, floor tiles, concrete roof tiles, Type S (ASTM C-144) perimeter security, pavers and traffic safety items such as concrete median barriers, bridge parapets, pedestrian 1 Part ½ Part 3½ - 4½ Parts crosswalks, curbs and other delineators. White cement is often Stucco Mix (Scratch & Brown ASTM C-926) used to produce bright finishes, vibrant colors or ultra high Washed Plaster performance concretes that look great and provide structural Hydrated Lime Portland Cement Sand performance that make them ideal for resilient building Type S applications. -

White Cement - Properties, Manufacture, Prospects
Review paper WHITE CEMENT - PROPERTIES, MANUFACTURE, PROSPECTS KARTEŘINA MORESOVÁ, FRANTIŠEK ŠKVÁRA Department of Glass and Ceramics, Institute of Chemical Technology, Technická 5, 166 28 Prague 6, Czech Republic E-mail: [email protected] Submitted January 6, 2000; accepted March 12, 2001. Keywords: White cement, Review, Properties INTRODUCTION stage and must also exhibit whiteness corresponding to cement of the given class. The raw materials, the Special cements differ from conventional Portland intermediate product and the final product must also be cements in their chemical and phase composition as protected from contamination at all stages of the well as in their properties. Their properties can be technology. The initial composition of the raw materials achieved by using a modified raw material mix, a or their properties are dealt with e.g. by [9 - 16]. grinding admixture or by adjusting the grinding fineness White cement may be manufactured with of the cement. Special cements include a number of standardized surface-active plasticizers or hydrophobic cements whose production volumes are considerably additives (in amounts of up to 0.5 wt.%) not impairing lower compared to those of conventional cements (max. the whiteness of the product. According to the 5 to 10 %). However, they constitute a significant part whiteness degree, white cement is divided into three of the assortment supplementing the conventional groups of the reflection coefficient: I. min. 80 %, II. cement. These materials include cements with high min. 75 % and III. min. 68 %. The whiteness degree is early strengths, sulphate-resistant Portland cements, determined according to the reflection coefficient in road cements, white cement and coloured cements, percent of the absolute scale. -

White CSA CEMENT Calcium Sulfo Aluminate
® white CSA CEMENT Calcium Sulfo Aluminate An innovation in cement technology for aesthetic and technical applications. is a special Calcium Sulfo Aluminate cement (CSA) designed for decorative concrete, terrazzo, flooring, glass fiber reinforced concrete (GFRC), dry-mixed mortars, architectural precast, fiber cement and more. Specially selected high purity raw materials, optimized calcination and carefully monitored grinding guarantee a consistent white color. Features: • Fast setting • High early strength (1-day strength equals the 28-day strength of ordinary cement) • Continuous strength development (no loss of strength in time) • Rapid drying (C4A3Š autogenously binds excess water) • Shrinkage compensated – (little to NO shrinkage) • Low alkali Benefits: • Ideal for making “ fast set concrete” • Allows for quick-demolding • Rapid return to service • Compatible with various aggregates • Reduces efflorescence Calcium Sulfoaluminate Cement increases strength, reduces set times, and decreases shrinkage of concrete mix designs. ,used as a stand-alone binder or blended with white portland cement delivers high early strength to incredibly durable concrete and mortar. Conventional retarding admixtures can be used to increase working time sacrificing early strength development Special Cement Department CALTRA US.- exclusive distributor; DELTA PERFORMANCE 9126 Industrial Blvd.,Suite B , Covington, GA 30014 T: +1 678 729 9330 F:+1 678 729 9336 M:+1 770 231 2674 W: www.caltra.com ® Calcium Sulfoaluminate Cement is ideal for applications requiring high early strength and rapid setting. Concrete and mortar formulated with CSA cement are capable of obtaining the 28-day strength of ordinary cement in just one day. Suitable projects include: • Concrete runway repair • Bridge deck repair • Tunneling • Roadway repairs • Non-shrink grout • Concrete floor overlayment • Concrete repair mortar Low Carbon Footprint The amount of energy used to produce Calcium Sulfoaluminate Cement is significantly lower than for portlandcement. -

Lehigh White Portland Limestone Cement ASTM 1157 GU
Lehigh White Portland Limestone Cement ASTM 1157 GU LEHIGH WHITE CEMENT PRODUCTS Lehigh White Cement Company is the Lehigh White Portland Cement makes leading supplier of white cements to the concrete brighter, better-looking and Portland Cement Type I concrete & pool plastering industry. environmentally friendly, without Lehigh White PLC Cement is backed by sacrificing the quality and special Portland Cement Type II/V our reputation for high-quality products attributes you expect from Lehigh that get the job done, every time. White Cement. Portland Limestone Cement Type GU RESPONSIBLE GREEN BUILDING Using White Portland Limestone Cement Portland Limestone Cement has been (PLC), is the smart and environmentally used internationally with great suc- responsible choice. This earth-friendly cess for more than 25 years, and is cement generates about 10% less CO2 now gaining popularity in North than Portland Cements, while delivering America with the increased emphasis great workability and the consistency on sustainable construction practices. that Lehigh White Cement Company Lehigh White Portland Limestone customers have relied upon for decades. Cement allows for the production of sustainable concrete products for PERFORMANCE owners and designers who want a Manufactured with the same care and green option in swimming pools and attention as all our cements, Lehigh architectural concrete products. White PLC meets ASTM C1157 Standard Performance Specification for Hydraulic AVAILABILITY Cement, Type GU and outperforms our Lehigh White Portland Limestone competitors’ Type I in concrete tests. Cement is available in the western United States including CA, NV, AZ USE and HI. To find out if Lehigh White Lehigh White Portland Limestone Cement PLC is available in your area or for can be used in almost any application sales assistance you can email us at where Portland Cement is normally used. -

White Portland Cement Material Safety Data Sheet
White Portland Cement Material Safety Data Sheet Section 1 - Identity Name: Argos White Portland Cement Address: 6505 Blue Lagoon dr. Miami Florida 33126 Date: October 2007 Emergency Telephone Number: 305 2670643 Common Name (used on label): Argos White Portland Cement Chemical Name: NONE Trade Name & Synonyms: WHITE PORTLAND CEMENT Chemical Family: MIXTURE OF CALCIUM SILICATES AND ALUMINUM SILICATE Formula: DOES NOT APPLY Section 2 - Hazardous Ingredients Hazardous Component CAS# % Typical TLV (Units) PEL (Units) PORTLAND CEMENT 65997-15-1 79 - 91 10 mg/m3 5 mg/m3* GYPSUM 13397-24-5 2 - 5 10 mg/m3 5 mg/m3* CALCIUM UNKNOWN 5 -12 2 mg/m3 5 mg/m3* HYDROXIDE *Respirable exposures. Total dust PELs are 15 mg/m3. **This material contains less than 1% crystalline silica. PEL: Permissible Exposure Limit established by the Occupational Safety and Health Administration (OSHA). TLV: Threshold Limit Value established by the American Conference of Governmental Industrial Hygienists (ACGIH). Section 3 - Physical Data Boiling Point Specific Gravity (H2O=1) Vapor Pressure (mm=Hg) DOES NOT APPLY 2.94 - 3.18 DOES NOT APPLY Evaporation Rate (n=Butyl Percent Volatile by Volume Vapor Density (Air=1) Acetate) 0% DOES NOT APPLY DOES NOT APPLY Percent Soluble in Water Reactivity in Water Appearance and Odor WILL NOT EVOLVE FINE, WHITE POWDER, NEGLIGIBLE (<0.1%) FLAMMABLE OR TOXIC NO ODOR GASES Hazardous Material Information System Identifier (HMIS) HEALTH = 2* FLAMMABILITY = 0 REACTIVITY = 0 PERSONAL PROTECTION = X *Indicates possibility of chronic health effects. Section 4 - Fire and Explosion Data Flash Point WILL NOT IGNITE Flammable Limits in Air (% by Volume) Lower: DOES NOT APPLY Upper: DOES NOT APPLY Extinguishing Media DOES NOT APPLY Auto Ignition Temperature DOES NOT APPLY Unusual Fire and Explosion Hazards NONE Special Fire Fighting Procedures NONE Section 5 - Health Information Signs and Symptoms of Exposure - (1) Acute Over exposure IRRITATION OF SKIN, EYES, NASAL PASSAGES AND RESPIRATORY SYSTEMS. -

The Durability of White Portland Cement to Chemical Attack
Downloaded from orbit.dtu.dk on: Dec 18, 2017 The durability of white Portland cement to chemical attack Nielsen, Erik Pram; Geiker, Mette Rica Publication date: 2004 Document Version Publisher's PDF, also known as Version of record Link back to DTU Orbit Citation (APA): Nielsen, E. P., & Geiker, M. R. (2004). The durability of white Portland cement to chemical attack. (BYG- Rapport; No. R-084). General rights Copyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. • Users may download and print one copy of any publication from the public portal for the purpose of private study or research. • You may not further distribute the material or use it for any profit-making activity or commercial gain • You may freely distribute the URL identifying the publication in the public portal If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. Erik Pram Nielsen The durability of white Portland cement to chemical attack DANMARKS TEKNISKE UNIVERSITET Report BYG·DTU R-084 2004 ISSN 1601-2917 ISBN 87-7877-147-1 SUMMARY In terms of volume, the severest conditions that reinforced concrete structures are subjected to usually involve some form of attack by chlorides. These include marine environments, roads and bridges exposed to de-icing salt, etc. The time it takes for the chlorides to reach the reinforcement is the main parameter used to predict the service life of the concrete structures. -

Castle White Portland Cement Complies with the Requirements
Hanson White Portland Cement Data Sheet Hanson White Portland Cement is manufactured to comply with the requirements of BS EN 197-1 CEM I Portland cement strength class 52,5. Hanson White Portland Cement is recommended for use in concrete, mortar and render with or without pigments. The setting and strength characteristics of Hanson White Portland Cement are similar to grey Portland cements. Hanson white cement is CE marked under the Construction Products Regulations which provides independent third party certification of product conformity. It confirms that in addition to applying a system of factory production control (defined in BS EN 197-2), independent sampling and testing of the cement has confirmed its compliance with all of the requirements of BS EN 197-1 Applications Hanson White Portland Cement is naturally chosen for its high degree of light reflection. To obtain white or light coloured concrete specially selected white or light coloured sand and stone aggregates are required. Coloured concretes and renders are achieved by adding pigments specifically designed for the use with Portland cement and Portland cement products. For extremely white concrete the white pigment Titanium Dioxide may be added in amounts of up to 5% by weight. Typical applications for Hanson White Portland Cement include: • cast stone • rendering • cladding panels • roadmarking, kerbs and bollards • exposed aggregate finishes • structural concrete • floor and wall tiles • swimming pools • pointing mortars • terrazzo finishes • precast units Quality Hanson White Portland Cement is manufactured from specially produced white Portland clinker using carefully selected raw materials avoiding minerals which would colour the final cement. The resultant clinker is ground in ceramic lined mills to ensure an excellent degree of whiteness. -

Safety Data Sheet (SDS) Section I. Identification Section II. Hazards
White Portland Cement Type I Page 1 of 9 Safety Data Sheet (SDS) Section I. Identification Product Identifier White Portland Cement Other Means of Identification White Portland Type I, White Portland Cement White Pool Cement, White Pool Cement Type I Chemical Name Calcium compounds, calcium silicate compounds, and other calcium compounds containing iron and aluminum make up most this product. Relevant Uses Building materials, construction application, a basic ingredient in concrete. Supplier Name SESCO Cement Corp. Address 8510 E Sam Houston Pkwy N Houston, TX 77044 Customer Care: 1-844-737-2687 Emergency Telephone Number CHEMTREC: 1-800-424-9300 Section II. Hazards Identification OSHA/HCS Status This material is considered hazardous by the OSHA Hazard Communication Standard (29 CFR 1910.1200). Category Classification(s) Skin Corrosion/Irritation - Category 1 Eye Damage - Category 1 Skin Sensitization - Category 1 Carcinogenicity/Inhalation - Category 1 Single Target Organ Toxicity (Repeated Exposure) - Category 2 GHS Label Elements: Hazard Pictograms GHS07 GHS05 GHS08 Signal Word Danger Hazard Statements Causes severe skin burns and eye damage May cause an allergic skin reaction May cause cancer (inhalation, dermal). Causes damage to lungs, kidneys and autoimmune system through prolonged or repeated exposure by inhalation Precautionary Statements: Prevention Obtain special instructions before use. Do not handle until all safety precautions have been read and understood. Do not breathe dust. Wash clothing, hands, forearms and face thoroughly after handling. Contaminated work clothing must not be allowed out of the workplace. Wear eye protection, protective clothing, protective gloves Response If swallowed: rinse mouth. Do NOT induce vomiting If on skin: Wash with plenty of soap and water If on skin (or hair): Take off immediately all contaminated clothing.