Development and Validation of a Quantitative Confirmatory Method for 30 Β-Lactam
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Drug Repositioning Case Study Prashant S. Kharkar1*, Ponnadurai Ramasami2, Yee S
Electronic Supplementary Material (ESI) for RSC Advances. This journal is © The Royal Society of Chemistry 2016 Discovery of Anti-Ebola Drugs: A Computational Drug Repositioning Case Study Prashant S. Kharkar1*, Ponnadurai Ramasami2, Yee Siew Choong3, Lydia Rhyman2 and Sona Warrier1 1SPP School of Pharmacy and Technology Management, SVKM’s NMIMS, V. L. Mehta Road, Vile Parle (West), Mumbai-400 056. INDIA. 2 Computational Chemistry Group, Department of Chemistry, Faculty of Science, University of Mauritius, Réduit 80837, Mauritius 3Institute for Research in Molecular Medicine (INFORMM), Universiti Sains Malaysia, Malaysia Contents Sr. No. Description Page No. 1 Table 1S 1 2 Figure 1S 28 3 Figure 2S 29 4 Figure 3S 30 5 Figure 4S 31 6 Figure 5S 32 Table 1S. Top hits (#100) identified from EON screening using query 1 Estimated Sr. EON_ Shape Free Energy Drug Structure ET_pb ET_coul ET_combo Rank No. Tanimoto of Binding (kcal/mol) OH O Br N O 1 1 S -9.88 HO O O O S O O 2 Sitaxentan S 0.633 0.914 0.926 0.294 1 -14.37 HN O Cl O N O HO 3 Alitretinoina 0.66 0.91 0.906 0.246 2 -3.91 NH 2 S N N O O HN 4 Ceftriaxone S 0.606 0.937 0.823 0.217 3 -12.33 N O S N O HO O N N O H O a 5 Acitretin O 0.637 0.936 0.809 0.172 4 -3.71 OH HO NH 2 HO N N 6 Cidofovir P O 0.473 0.633 0.783 0.31 5 -4.21 HO O O N N N N 7 Telmisartan 0.601 0.908 0.775 0.174 6 -6.46 OH O 8 Nateglinidea HN 0.54 0.874 0.745 0.205 7 -3.78 OH O O H N 2 S OH N O N S O 9 Ceftizoxime N 0.557 0.88 0.735 0.178 8 -11.35 H O N O OH O 10 Treprostinil OH 0.432 0.846 0.732 0.301 9 -3.41 O HO O O S -

AMEG Categorisation of Antibiotics
12 December 2019 EMA/CVMP/CHMP/682198/2017 Committee for Medicinal Products for Veterinary use (CVMP) Committee for Medicinal Products for Human Use (CHMP) Categorisation of antibiotics in the European Union Answer to the request from the European Commission for updating the scientific advice on the impact on public health and animal health of the use of antibiotics in animals Agreed by the Antimicrobial Advice ad hoc Expert Group (AMEG) 29 October 2018 Adopted by the CVMP for release for consultation 24 January 2019 Adopted by the CHMP for release for consultation 31 January 2019 Start of public consultation 5 February 2019 End of consultation (deadline for comments) 30 April 2019 Agreed by the Antimicrobial Advice ad hoc Expert Group (AMEG) 19 November 2019 Adopted by the CVMP 5 December 2019 Adopted by the CHMP 12 December 2019 Official address Domenico Scarlattilaan 6 ● 1083 HS Amsterdam ● The Netherlands Address for visits and deliveries Refer to www.ema.europa.eu/how-to-find-us Send us a question Go to www.ema.europa.eu/contact Telephone +31 (0)88 781 6000 An agency of the European Union © European Medicines Agency, 2020. Reproduction is authorised provided the source is acknowledged. Categorisation of antibiotics in the European Union Table of Contents 1. Summary assessment and recommendations .......................................... 3 2. Introduction ............................................................................................ 7 2.1. Background ........................................................................................................ -

Consideration of Antibacterial Medicines As Part Of
Consideration of antibacterial medicines as part of the revisions to 2019 WHO Model List of Essential Medicines for adults (EML) and Model List of Essential Medicines for children (EMLc) Section 6.2 Antibacterials including Access, Watch and Reserve Lists of antibiotics This summary has been prepared by the Health Technologies and Pharmaceuticals (HTP) programme at the WHO Regional Office for Europe. It is intended to communicate changes to the 2019 WHO Model List of Essential Medicines for adults (EML) and Model List of Essential Medicines for children (EMLc) to national counterparts involved in the evidence-based selection of medicines for inclusion in national essential medicines lists (NEMLs), lists of medicines for inclusion in reimbursement programs, and medicine formularies for use in primary, secondary and tertiary care. This document does not replace the full report of the WHO Expert Committee on Selection and Use of Essential Medicines (see The selection and use of essential medicines: report of the WHO Expert Committee on Selection and Use of Essential Medicines, 2019 (including the 21st WHO Model List of Essential Medicines and the 7th WHO Model List of Essential Medicines for Children). Geneva: World Health Organization; 2019 (WHO Technical Report Series, No. 1021). Licence: CC BY-NC-SA 3.0 IGO: https://apps.who.int/iris/bitstream/handle/10665/330668/9789241210300-eng.pdf?ua=1) and Corrigenda (March 2020) – TRS1021 (https://www.who.int/medicines/publications/essentialmedicines/TRS1021_corrigenda_March2020. pdf?ua=1). Executive summary of the report: https://apps.who.int/iris/bitstream/handle/10665/325773/WHO- MVP-EMP-IAU-2019.05-eng.pdf?ua=1. -

Computational Antibiotics Book
Andrew V DeLong, Jared C Harris, Brittany S Larcart, Chandler B Massey, Chelsie D Northcutt, Somuayiro N Nwokike, Oscar A Otieno, Harsh M Patel, Mehulkumar P Patel, Pratik Pravin Patel, Eugene I Rowell, Brandon M Rush, Marc-Edwin G Saint-Louis, Amy M Vardeman, Felicia N Woods, Giso Abadi, Thomas J. Manning Computational Antibiotics Valdosta State University is located in South Georgia. Computational Antibiotics Index • Computational Details and Website Access (p. 8) • Acknowledgements (p. 9) • Dedications (p. 11) • Antibiotic Historical Introduction (p. 13) Introduction to Antibiotic groups • Penicillin’s (p. 21) • Carbapenems (p. 22) • Oxazolidines (p. 23) • Rifamycin (p. 24) • Lincosamides (p. 25) • Quinolones (p. 26) • Polypeptides antibiotics (p. 27) • Glycopeptide Antibiotics (p. 28) • Sulfonamides (p. 29) • Lipoglycopeptides (p. 30) • First Generation Cephalosporins (p. 31) • Cephalosporin Third Generation (p. 32) • Fourth-Generation Cephalosporins (p. 33) • Fifth Generation Cephalosporin’s (p. 34) • Tetracycline antibiotics (p. 35) Computational Antibiotics Antibiotics Covered (in alphabetical order) Amikacin (p. 36) Cefempidone (p. 98) Ceftizoxime (p. 159) Amoxicillin (p. 38) Cefepime (p. 100) Ceftobiprole (p. 161) Ampicillin (p. 40) Cefetamet (p. 102) Ceftoxide (p. 163) Arsphenamine (p. 42) Cefetrizole (p. 104) Ceftriaxone (p. 165) Azithromycin (p.44) Cefivitril (p. 106) Cefuracetime (p. 167) Aziocillin (p. 46) Cefixime (p. 108) Cefuroxime (p. 169) Aztreonam (p.48) Cefmatilen ( p. 110) Cefuzonam (p. 171) Bacampicillin (p. 50) Cefmetazole (p. 112) Cefalexin (p. 173) Bacitracin (p. 52) Cefodizime (p. 114) Chloramphenicol (p.175) Balofloxacin (p. 54) Cefonicid (p. 116) Cilastatin (p. 177) Carbenicillin (p. 56) Cefoperazone (p. 118) Ciprofloxacin (p. 179) Cefacetrile (p. 58) Cefoselis (p. 120) Clarithromycin (p. 181) Cefaclor (p. -

Información Del Principio Activo
Subscribe to DeepL Pro to edit this document. Visit www.DeepL.com/Pro for more information. SERVINSUMOS S.A. LABORATORIES TECHNICAL SHEET CEFTISER Code: FT- 67 Date: 17-02-2010 Version: 1 Page: 1 of 3 ACTIVE INGREDIENT INFORMATION 1.1 Molecule Ceftiofur HCl 1.2 Generic Name Ceftiofur 1.3 Empirical Formula: C19H17N5O7S3- 1.4 Structural Formula HCl PRODUCT INFORMATION 2.1 Trade name CEFTISER 2.2 Composition Each mL of solution contains: Ceftiofur clorhidrato 50 mg Excipients c.s.p. 1 mL 2.3 Pharmaceutical Form Injectable Solution 2.4 Species Cattle, pigs, horses, sheep, goats and dogs. 2.5 Indications Ceftiofur is a third generation cephalosporin with a broad spectrum, indicated in cattle, swine, horses, sheep, goats and dogs for the treatment and control of infectious diseases produced by Pasteurella haemolytica, Pasteurella multocida, Actinobacillus pleuropneumoniae (respiratory infections), Streptococcus s.p, Staphylococcus s.p, (respiratory tract infections, metritis where these bacteria are involved), Escherichia coli and Salmonella s.p (Infections of the respiratory and digestive tract), Haemophillus s.p (infectious polyarthritis and respiratory infection in the pig), Fusobacterium necrophorum (necrobacillosis or bovine pododermatitis). Its effectiveness in the treatment of respiratory diseases and in the treatment of necrobacillosis or hoof rot in cattle, sheep and goats is highlighted. TECHNICAL SHEET CEFTISER Code: FT- 67 Date: 17-02-2010 Version: 1 Page 2of 3 2.6 Administration Intramuscular or subcutaneous route. 2.7 Dose Recommended dosage: In cattle, sheep, goats: Administer by intramuscular or subcutaneous route 1 - 2 mg/Kg of weight every 24 hours. In the practice 1 to 2 mL for each 50 Kg of live weight, daily during three days In pigs: To administer by intramuscular route 3 to 5 mg/kg of weight each 24 hours. -

European Surveillance of Healthcare-Associated Infections in Intensive Care Units
TECHNICAL DOCUMENT European surveillance of healthcare-associated infections in intensive care units HAI-Net ICU protocol Protocol version 1.02 www.ecdc.europa.eu ECDC TECHNICAL DOCUMENT European surveillance of healthcare- associated infections in intensive care units HAI-Net ICU protocol, version 1.02 This technical document of the European Centre for Disease Prevention and Control (ECDC) was coordinated by Carl Suetens. In accordance with the Staff Regulations for Officials and Conditions of Employment of Other Servants of the European Union and the ECDC Independence Policy, ECDC staff members shall not, in the performance of their duties, deal with a matter in which, directly or indirectly, they have any personal interest such as to impair their independence. This is version 1.02 of the HAI-Net ICU protocol. Differences between versions 1.01 (December 2010) and 1.02 are purely editorial. Suggested citation: European Centre for Disease Prevention and Control. European surveillance of healthcare- associated infections in intensive care units – HAI-Net ICU protocol, version 1.02. Stockholm: ECDC; 2015. Stockholm, March 2015 ISBN 978-92-9193-627-4 doi 10.2900/371526 Catalogue number TQ-04-15-186-EN-N © European Centre for Disease Prevention and Control, 2015 Reproduction is authorised, provided the source is acknowledged. TECHNICAL DOCUMENT HAI-Net ICU protocol, version 1.02 Table of contents Abbreviations ............................................................................................................................................... -

Directed Molecular Evolution of Fourth-Generation Cephalosporin Resistance in Wellington Moore Iowa State University
Iowa State University Capstones, Theses and Graduate Theses and Dissertations Dissertations 2011 Directed molecular evolution of fourth-generation cephalosporin resistance in Wellington Moore Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/etd Part of the Medical Sciences Commons Recommended Citation Moore, Wellington, "Directed molecular evolution of fourth-generation cephalosporin resistance in" (2011). Graduate Theses and Dissertations. 10107. https://lib.dr.iastate.edu/etd/10107 This Thesis is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Graduate Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. Directed molecular evolution of fourth-generation cephalosporin resistance in Salmonella and Yersinia by Wellington Moore A thesis submitted to the graduate faculty in partial fulfillment of the requirements for the degree of MASTER OF SCIENCE Major: Biomedical Science (Pharmacology) Program of Study Committee: Steve Carlson, Major Professor Timothy Day Ronald Griffith Iowa State University Ames, Iowa 2011 ii TABLE OF CONTENTS LIST OF FIGURES………………………………………………………………………iii LIST OF TABLES………………………………………………………………………..iv ABSTRACT……………………………………………………………………………….v CHAPTER 1. INTRODUCTION…………………………………………………………1 Review of B-Lactam antimicrobials………………………………...………1 -
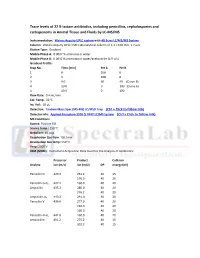
Trace Levels of 22 ß-Lactam Antibiotics, Including Penicillins, Cephalosporins and Carbapenems in Animal Tissue and Fluids by LC-MS/MS
Trace levels of 22 ß-lactam antibiotics, including penicillins, cephalosporins and carbapenems in Animal Tissue and Fluids by LC-MS/MS Instrumentation: Waters Acquity UPLC system with AB Sciex LC/MS/MS System Column: Waters Acquity UPLC CSH C18 analytical column of 2.1 × 100 mm, 1.7 µm Elution Type: Gradient Mobile Phase A: 0.0032 % ammonia in water Mobile Phase B: 0.0032 % ammonia in water/acetonitrile (1:9 v/v) Gradient Profile: Step No. Time (min) Pct A Pct B 1 0 100 0 2 1 100 0 3 9.0 60 40 (Curve 6) 4 10.0 0 100 (Curve 6) 4 10.5 0 100 Flow Rate: 0.4 mL/min Col. Temp: 50 °C Inj. Vol.: 10 µL Detection: Tandem Mass Spec (MS-MS) LC/MSD Trap (Ctrl + Click to follow link) Detector Info: Applied Biosystem 3200 Q TRAP LC/MS System (Ctrl + Click to follow link) MS Conditions Source: Positive ESI Source temp.: 150 °C Nebulizer: 40 psig Desolvation Gas flow: 10 L/min Desolvation Gas temp: 550 °C Vcap: 2000 V SRM (MRM): Instrument Acquisition Data Used for the Analysis of Antibiotics: Precursor Product Collision Analyte ion (m/z) ion (m/z) DP energy (eV) Penicillin G 420.2 261.2 40 15 176.0 40 20 Penicillin G-d7 427.2 160.0 40 20 Ampicillin 435.2 286.0 40 20 276.2 40 20 Ampicillin-d5 440.2 291.0 40 20 Penicillin V 436.0 277.0 40 20 192.0 40 20 160.0 40 20 Penicillin-V-d5 441.0 160.0 40 20 Amoxicillin 451.2 275.2 40 15 302.2 40 15 SRM (MRM): Instrument Acquisition Data Used for the Analysis of Antibiotics (cont.): Precursor Product Collision Analyte ion (m/z) ion (m/z) DP energy (eV) Amoxicillin-d4 455.2 279.2 40 15 Oxacillin 487.0 160.0 40 17 -

Advances in Antimicrobial Resistance Monitoring Using Sensors and Biosensors: a Review
chemosensors Review Advances in Antimicrobial Resistance Monitoring Using Sensors and Biosensors: A Review Eduardo C. Reynoso 1 , Serena Laschi 2, Ilaria Palchetti 3,* and Eduardo Torres 1,4 1 Ciencias Ambientales, Instituto de Ciencias, Benemérita Universidad Autónoma de Puebla, Puebla 72570, Mexico; [email protected] (E.C.R.); [email protected] (E.T.) 2 Nanobiosens Join Lab, Università degli Studi di Firenze, Viale Pieraccini 6, 50139 Firenze, Italy; [email protected] 3 Dipartimento di Chimica, Università degli Studi di Firenze, Via della Lastruccia 3, 50019 Sesto Fiorentino, Italy 4 Centro de Quìmica, Benemérita Universidad Autónoma de Puebla, Puebla 72570, Mexico * Correspondence: ilaria.palchetti@unifi.it Abstract: The indiscriminate use and mismanagement of antibiotics over the last eight decades have led to one of the main challenges humanity will have to face in the next twenty years in terms of public health and economy, i.e., antimicrobial resistance. One of the key approaches to tackling an- timicrobial resistance is clinical, livestock, and environmental surveillance applying methods capable of effectively identifying antimicrobial non-susceptibility as well as genes that promote resistance. Current clinical laboratory practices involve conventional culture-based antibiotic susceptibility testing (AST) methods, taking over 24 h to find out which medication should be prescribed to treat the infection. Although there are techniques that provide rapid resistance detection, it is necessary to have new tools that are easy to operate, are robust, sensitive, specific, and inexpensive. Chemical sensors and biosensors are devices that could have the necessary characteristics for the rapid diag- Citation: Reynoso, E.C.; Laschi, S.; nosis of resistant microorganisms and could provide crucial information on the choice of antibiotic Palchetti, I.; Torres, E. -

Validation of a Rapid Lateral Flow Test for the Simultaneous Determination of B-Lactam Drugs and Flunixin in Raw Milk
® Validation of a Rapid Lateral Flow Test for the Simultaneous Determination of b-Lactam Drugs and Flunixin in Raw Milk DAVID DOUGLAS,1 KATIE BANASZEWSKI,2 RIMA JUSKELIS,2 FADWA AL-TAHER,2 YANG CHEN,2 JACK CAPPOZZO,2 LINDSAY MCROBBIE,1 AND ROBERT S. SALTER1* 1Charm Sciences, Inc., 659 Andover Street, Lawrence, Massachusetts 01843; and 2Institute for Food Safety and Health–National Center for Food Safety and Technology at the Illinois Institute of Technology, 6502 South Archer Road, Bedford Park, Illinois 60501, USA 1270 Journal of Food Protection, Vol. 75, No. 7, 2012, Pages 1270–1277 doi:10.4315/0362-028X.JFP-11-570 Copyright G, International Association for Food Protection Validation of a Rapid Lateral Flow Test for the Simultaneous Determination of b-Lactam Drugs and Flunixin in Raw Milk DAVID DOUGLAS,1 KATIE BANASZEWSKI,2 RIMA JUSKELIS,2 FADWA AL-TAHER,2 YANG CHEN,2 JACK CAPPOZZO,2 LINDSAY MCROBBIE,1 AND ROBERT S. SALTER1* 1Charm Sciences, Inc., 659 Andover Street, Lawrence, Massachusetts 01843; and 2Institute for Food Safety and Health–National Center for Food Safety and Technology at the Illinois Institute of Technology, 6502 South Archer Road, Bedford Park, Illinois 60501, USA MS 11-570: Received 30 December 2011/Accepted 17 March 2012 ABSTRACT b-Lactam antibiotics are the most commonly used drugs on dairy farms. b-Lactam residues in milk are kept out of the human milk supply with good agricultural practices and mandatory truck screening performed by the dairy industry under Appendix N of the Pasteurized Milk Ordinance. Flunixin, a nonsteroidal and anti-inflammatory drug, appears in dairy cattle tissue residues with a frequency similar to the occurrence of penicillin G. -

Antibiotic Classes
Penicillins Aminoglycosides Generic Brand Name Generic Brand Name Amoxicillin Amoxil, Polymox, Trimox, Wymox Amikacin Amikin Ampicillin Omnipen, Polycillin, Polycillin-N, Gentamicin Garamycin, G-Mycin, Jenamicin Principen, Totacillin, Unasyn Kanamycin Kantrex Bacampicillin Spectrobid Neomycin Mycifradin, Myciguent Carbenicillin Geocillin, Geopen Netilmicin Netromycin Cloxacillin Cloxapen Paromomycin Dicloxacillin Dynapen, Dycill, Pathocil Streptomycin Flucloxacillin Flopen, Floxapen, Staphcillin Tobramycin Nebcin Mezlocillin Mezlin Nafcillin Nafcil, Nallpen, Unipen Quinolones Oxacillin Bactocill, Prostaphlin Generic Brand Name Penicillin G Bicillin L-A, First Generation Crysticillin 300 A.S., Pentids, Flumequine Flubactin Permapen, Pfizerpen, Pfizerpen- Nalidixic acid NegGam, Wintomylon AS, Wycillin Oxolinic acid Uroxin Penicillin V Beepen-VK, Betapen-VK, Piromidic acid Panacid Ledercillin VK, V-Cillin K Pipemidic acid Dolcol Piperacillin Pipracil, Zosyn Rosoxacin Eradacil Pivampicillin Second Generation Pivmecillinam Ciprofloxacin Cipro, Cipro XR, Ciprobay, Ciproxin Ticarcillin Ticar Enoxacin Enroxil, Penetrex Lomefloxacin Maxaquin Monobactams Nadifloxacin Acuatim, Nadoxin, Nadixa Generic Brand Name Norfloxacin Lexinor, Noroxin, Quinabic, Aztreonam Azactam, Cayston Janacin Ofloxacin Floxin, Oxaldin, Tarivid Carbapenems Pefloxacin Peflacine Generic Brand Name Rufloxacin Uroflox Imipenem, Primaxin Third Generation Imipenem/cilastatin Balofloxacin Baloxin Doripenem Doribax Gatifloxacin Tequin, Zymar Meropenem Merrem Grepafloxacin Raxar Ertapenem -

One Analysis, One Column, Less Than 9 Minutes for Over 60 Multiclass Antibiotics
Featured Application: Multiclass Veterinary Antibiotics on Raptor C18 by LC-MS/MS One Analysis, One Column, Less than 9 Minutes for Over 60 Multiclass Antibiotics • Highly efficient peak separation and fast analysis times. • Versatility and global applicability for antibiotic residue analysis—capable of individual class panel optimization for quantitation: - Macrolide, Lincosamide, and Streptogramin (Figure 1) - Amphenicol and Tetracycline (Figure 2) - Quinolone (Figure 3) - Penicillin, Cephalosporin, and Tetracycline (Figure 4) - Sulfonamide (Figure 5) (For Ionophore, use on Raptor Biphenyl. [Figure 6]) The use of antibiotics on food-producing animals is a public health and safety concern due to the potential of generating drug-resistant bacteria. Many countries in the European Union and Canada have banned the use of antibiotics for nontherapeutic purposes, and the United States is implementing a policy to reduce the use of medically important antibiotics for growth promotion. To regulate the proper use of veterinary antibiotics, the U.S. FDA has set maximum residue lim- its (MRL) for a variety of animal tissue and food products (21 CFR Part 556). A sensitive, efficient, and reliable analytical method for different classes of antibiotics is necessary to meet this regulation, and the Raptor C18 LC column is the ideal choice. Conc. Conc. Peaks tR (min) (ng/mL) Precursor Ion Product Ion Product Ion Peaks tR (min) (ng/mL) Precursor Ion Product Ion Product Ion 1. Desacetyl cephapirin 0.70 150 382.03 111.92 124.21 31. Sarafloxacin 2.59 10 386.20 342.20 368.15 2. Sulfanilamide 0.85 200 172.98 93.07 75.23 32. Difloxacin 2.65 10 400.23 356.17 299.13 3.