82010795.Pdf
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Ion Channels
UC Davis UC Davis Previously Published Works Title THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Ion channels. Permalink https://escholarship.org/uc/item/1442g5hg Journal British journal of pharmacology, 176 Suppl 1(S1) ISSN 0007-1188 Authors Alexander, Stephen PH Mathie, Alistair Peters, John A et al. Publication Date 2019-12-01 DOI 10.1111/bph.14749 License https://creativecommons.org/licenses/by/4.0/ 4.0 Peer reviewed eScholarship.org Powered by the California Digital Library University of California S.P.H. Alexander et al. The Concise Guide to PHARMACOLOGY 2019/20: Ion channels. British Journal of Pharmacology (2019) 176, S142–S228 THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Ion channels Stephen PH Alexander1 , Alistair Mathie2 ,JohnAPeters3 , Emma L Veale2 , Jörg Striessnig4 , Eamonn Kelly5, Jane F Armstrong6 , Elena Faccenda6 ,SimonDHarding6 ,AdamJPawson6 , Joanna L Sharman6 , Christopher Southan6 , Jamie A Davies6 and CGTP Collaborators 1School of Life Sciences, University of Nottingham Medical School, Nottingham, NG7 2UH, UK 2Medway School of Pharmacy, The Universities of Greenwich and Kent at Medway, Anson Building, Central Avenue, Chatham Maritime, Chatham, Kent, ME4 4TB, UK 3Neuroscience Division, Medical Education Institute, Ninewells Hospital and Medical School, University of Dundee, Dundee, DD1 9SY, UK 4Pharmacology and Toxicology, Institute of Pharmacy, University of Innsbruck, A-6020 Innsbruck, Austria 5School of Physiology, Pharmacology and Neuroscience, University of Bristol, Bristol, BS8 1TD, UK 6Centre for Discovery Brain Science, University of Edinburgh, Edinburgh, EH8 9XD, UK Abstract The Concise Guide to PHARMACOLOGY 2019/20 is the fourth in this series of biennial publications. The Concise Guide provides concise overviews of the key properties of nearly 1800 human drug targets with an emphasis on selective pharmacology (where available), plus links to the open access knowledgebase source of drug targets and their ligands (www.guidetopharmacology.org), which provides more detailed views of target and ligand properties. -

Replicated Risk Nicotinic Cholinergic Receptor Genes for Nicotine Dependence
G C A T T A C G G C A T genes Article Replicated Risk Nicotinic Cholinergic Receptor Genes for Nicotine Dependence Lingjun Zuo 1, Rolando Garcia-Milian 2, Xiaoyun Guo 1,3,4,*, Chunlong Zhong 5,*, Yunlong Tan 6, Zhiren Wang 6, Jijun Wang 3, Xiaoping Wang 7, Longli Kang 8, Lu Lu 9,10, Xiangning Chen 11,12, Chiang-Shan R. Li 1 and Xingguang Luo 1,6,* 1 Department of Psychiatry, Yale University School of Medicine, New Haven, CT 06510, USA; [email protected] (L.Z.); [email protected] (C.-S.R.L.) 2 Curriculum & Research Support Department, Cushing/Whitney Medical Library, Yale University School of Medicine, New Haven, CT 06510, USA; [email protected] 3 Shanghai Mental Health Center, Shanghai 200030, China; [email protected] 4 Department of Cellular and Molecular Physiology, Yale University School of Medicine, New Haven, CT 06510, USA 5 Department of Neurosurgery, Ren Ji Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai 200127, China 6 Biological Psychiatry Research Center, Beijing Huilongguan Hospital, Beijing 100096, China; [email protected] (Y.T.); [email protected] (Z.W.) 7 Department of Neurology, Shanghai First People’s Hospital, Shanghai Jiao Tong University, Shanghai 200080, China; [email protected] 8 Key Laboratory for Molecular Genetic Mechanisms and Intervention Research on High Altitude Diseases of Tibet Autonomous Region, Xizang Minzu University School of Medicine, Xianyang, Shanxi 712082, China; [email protected] 9 Provincial Key Laboratory for Inflammation and Molecular Drug Target, Medical -

Nicotinic Receptors in Sleep-Related Hypermotor Epilepsy: Pathophysiology and Pharmacology
brain sciences Review Nicotinic Receptors in Sleep-Related Hypermotor Epilepsy: Pathophysiology and Pharmacology Andrea Becchetti 1,* , Laura Clara Grandi 1 , Giulia Colombo 1 , Simone Meneghini 1 and Alida Amadeo 2 1 Department of Biotechnology and Biosciences, University of Milano-Bicocca, 20126 Milano, Italy; [email protected] (L.C.G.); [email protected] (G.C.); [email protected] (S.M.) 2 Department of Biosciences, University of Milano, 20133 Milano, Italy; [email protected] * Correspondence: [email protected] Received: 13 October 2020; Accepted: 21 November 2020; Published: 25 November 2020 Abstract: Sleep-related hypermotor epilepsy (SHE) is characterized by hyperkinetic focal seizures, mainly arising in the neocortex during non-rapid eye movements (NREM) sleep. The familial form is autosomal dominant SHE (ADSHE), which can be caused by mutations in genes encoding subunits of the neuronal nicotinic acetylcholine receptor (nAChR), Na+-gated K+ channels, as well as non-channel signaling proteins, such as components of the gap activity toward rags 1 (GATOR1) macromolecular complex. The causative genes may have different roles in developing and mature brains. Under this respect, nicotinic receptors are paradigmatic, as different pathophysiological roles are exerted by distinct nAChR subunits in adult and developing brains. The widest evidence concerns α4 and β2 subunits. These participate in heteromeric nAChRs that are major modulators of excitability in mature neocortical circuits as well as regulate postnatal synaptogenesis. However, growing evidence implicates mutant α2 subunits in ADSHE, which poses interpretive difficulties as very little is known about the function of α2-containing (α2*) nAChRs in the human brain. -

Structural Analysis of Pathogenic Missense Mutations in GABRA2 and Identification of a Novel De Novo Variant in the Desensitization Gate
Received: 22 October 2019 | Revised: 29 November 2019 | Accepted: 10 December 2019 DOI: 10.1002/mgg3.1106 ORIGINAL ARTICLE Structural analysis of pathogenic missense mutations in GABRA2 and identification of a novel de novo variant in the desensitization gate Alba Sanchis-Juan1,2 | Marcia A. Hasenahuer3,4 | James A. Baker3 | Amy McTague5 | Katy Barwick5 | Manju A. Kurian5 | Sofia T. Duarte6 | NIHR BioResource | Keren J. Carss1,2 | Janet Thornton3 | F. Lucy Raymond2,4 1Department of Haematology, University of Cambridge, NHS Blood and Transplant Abstract Centre, Cambridge, UK Background: Cys-loop receptors control neuronal excitability in the brain and their 2NIHR BioResource, Cambridge dysfunction results in numerous neurological disorders. Recently, six missense vari- University Hospitals NHS Foundation ants in GABRA2, a member of this family, have been associated with early infantile Trust, Cambridge Biomedical Campus, Cambridge, UK epileptic encephalopathy (EIEE). We identified a novel de novo missense variant 3European Molecular Biology Laboratory, in GABRA2 in a patient with EIEE and performed protein structural analysis of the European Bioinformatics Institute, seven variants. Wellcome Genome Campus, Hinxton, . Cambridge, UK Methods: The novel variant was identified by trio whole-genome sequencing We 4Department of Medical Genetics, performed protein structural analysis of the seven variants, and compared them to Cambridge Institute for Medical Research, previously reported pathogenic mutations at equivalent positions in other Cys-loop University of Cambridge, Cambridge, UK receptors. Additionally, we studied the distribution of disease-associated variants in 5Developmental Neurosciences, Great the transmembrane helices of these proteins. Ormond Street Institute of Child Health, University College London, London, UK Results: The seven variants are in the transmembrane domain, either close to the de- 6Hospital Dona Estefânia, Centro Hospitalar sensitization gate, the activation gate, or in inter-subunit interfaces. -

Expression of Nicotinic Acetylcholine Receptor Subunit Genes in Non–Small-Cell Lung Cancer Reveals Differences Between Smokers and Nonsmokers
Research Article Expression of Nicotinic Acetylcholine Receptor Subunit Genes in Non–Small-Cell Lung Cancer Reveals Differences between Smokers and Nonsmokers David Chi-leung Lam,1,2 Luc Girard,4 Ruben Ramirez,4 Wing-shun Chau,3 Wai-sing Suen,3 Shelley Sheridan,4 Vicky P.C. Tin,2 Lap-ping Chung,2 Maria P. Wong,2 Jerry W. Shay,5 Adi F. Gazdar,4 Wah-kit Lam,1 and John D. Minna4 Departments of 1Medicine and 2Pathology, University of Hong Kong; 3Cardiothoracic Surgical Unit, The Grantham Hospital, HKSAR, China; and 4Hamon Center for Therapeutic Oncology Research and 5Department of Cell Biology, Simmons Comprehensive Cancer Center, University of Texas Southwestern Medical Center at Dallas, Dallas, Texas Abstract could also participate in lung cancer pathogenesis by activating Nicotine and its derivatives, by binding to nicotinic acetyl- signal transduction pathways such as the Akt pathway (1). One choline receptors (nAChR) on bronchial epithelial cells, can model could be that nicotine by stimulating nicotinic AChRs regulate cellular proliferation and apoptosis via activating the (nAChR) would activate Akt in lung epithelial cells and perhaps Akt pathway. Delineation of nAChR subtypes in non–small-cell stimulate cell proliferation and/or overcome apoptotic responses lung cancers (NSCLC) may provide information for prevention engendered by carcinogen exposure (1). If this model is true, then or therapeutic targeting. Expression of nAChR subunit genes one may ask whether lung tumors have different nAChR expression in 66 resected primary NSCLCs, 7 histologically non-involved patterns compared with normal lung tissues and whether lung lung tissues, 13 NSCLC cell lines, and 6 human bronchial cancers arising in smokers have different patterns compared with epithelial cell lines (HBEC) was analyzed with quantitative never smokers. -

Resequencing of Nicotinic Acetylcholine Receptor Genes and Association of Common and Rare Variants with the Fagerstro¨M Test for Nicotine Dependence
Neuropsychopharmacology (2010) 35, 2392–2402 & 2010 Nature Publishing Group All rights reserved 0893-133X/10 $32.00 www.neuropsychopharmacology.org Resequencing of Nicotinic Acetylcholine Receptor Genes and Association of Common and Rare Variants with the Fagerstro¨m Test for Nicotine Dependence 1,4 1 2 2 1 Jennifer Wessel , Sarah M McDonald , David A Hinds , Renee P Stokowski , Harold S Javitz , 2 1 2 1 2 1 Michael Kennemer , Ruth Krasnow , William Dirks , Jill Hardin , Steven J Pitts , Martha Michel , 1 2 3 1 ,1 Lisa Jack , Dennis G Ballinger , Jennifer B McClure , Gary E Swan and Andrew W Bergen* 1 2 3 Center for Health Sciences, SRI International, Menlo Park, CA, USA; Perlegen Sciences, Mountain View, CA, USA; Group Health Research 4 Institute, Seattle, WA, USA; Department of Public Health, Indiana University School of Medicine, Indianapolis, IN, USA Common single-nucleotide polymorphisms (SNPs) at nicotinic acetylcholine receptor (nAChR) subunit genes have previously been associated with measures of nicotine dependence. We investigated the contribution of common SNPs and rare single-nucleotide variants (SNVs) in nAChR genes to Fagerstro¨m test for nicotine dependence (FTND) scores in treatment-seeking smokers. Exons of 10 genes were resequenced with next-generation sequencing technology in 448 European-American participants of a smoking cessation trial, and CHRNB2 and CHRNA4 were resequenced by Sanger technology to improve sequence coverage. A total of 214 SNP/SNVs were identified, of which 19.2% were excluded from analyses because of reduced completion rate, 73.9% had minor allele frequencies o5%, and 48.1% were novel relative to dbSNP build 129. -

Lung Cancer and Nicotine
aphy & S gr ep to a a ra t m i o o r n h T C e Bharti and Yashila, J Chromatogr Sep Tech 2016, 7:2 f c o h Journal of Chromatography l n a i DOI: 10.4172/2157-7064.1000319 q n u r e u s o J Separation Techniques ISSN: 2157-7064 Review Article OpenOpen Access Access Lung Cancer and Nicotine Bharti M1* and Yashila G2 1National Institute of Pharmaceutical Education and Research, Mohali, Punjab-160 062, India 2Department of Biotechnology, Thapar University, Patiala, Punjab-147 002, India Abstract Nicotine present in smoking and tobacco is major cause of occurrence of lung cancer. The basic nature of nicotine helps in easy absorption through lungs. The binding of nicotine and its derivatives to Nicotinic Acetylcholine Receptor results in significant polymorphic mutations in genes coding the subunits of receptors in various populations like Asian and Caucasians which increases the susceptibility of lung cancer in these populations. Keywords: Nicotine; Lung cancer; Nicotine acetylcholine receptors competitively to the Nicotine Acetylcholine Receptor (nAChRs) present in brain as well as in lungs [6]. Nicotine is get metabolized in Introduction liver along with lungs and kidneys and responsible for the production Lung cancer is the third most common cancer after prostate gland of highly carcinogenic intermediates and by-products which also and breast cancer. A study has been reported 63,000 deaths per year in annex to the Nicotine Acetylcholine Receptor (nAChRs) and cause India due to the lung cancer [1]. Although, there must be numerous alterations in the receptors. -

Ligand-Gated Ion Channels
S.P.H. Alexander et al. The Concise Guide to PHARMACOLOGY 2015/16: Ligand-gated ion channels. British Journal of Pharmacology (2015) 172, 5870–5903 THE CONCISE GUIDE TO PHARMACOLOGY 2015/16: Ligand-gated ion channels Stephen PH Alexander1, John A Peters2, Eamonn Kelly3, Neil Marrion3, Helen E Benson4, Elena Faccenda4, Adam J Pawson4, Joanna L Sharman4, Christopher Southan4, Jamie A Davies4 and CGTP Collaborators L 1 School of Biomedical Sciences, University of Nottingham Medical School, Nottingham, NG7 2UH, UK, N 2Neuroscience Division, Medical Education Institute, Ninewells Hospital and Medical School, University of Dundee, Dundee, DD1 9SY, UK, 3School of Physiology and Pharmacology, University of Bristol, Bristol, BS8 1TD, UK, 4Centre for Integrative Physiology, University of Edinburgh, Edinburgh, EH8 9XD, UK Abstract The Concise Guide to PHARMACOLOGY 2015/16 provides concise overviews of the key properties of over 1750 human drug targets with their pharmacology, plus links to an open access knowledgebase of drug targets and their ligands (www.guidetopharmacology.org), which provides more detailed views of target and ligand properties. The full contents can be found at http://onlinelibrary.wiley.com/ doi/10.1111/bph.13350/full. Ligand-gated ion channels are one of the eight major pharmacological targets into which the Guide is divided, with the others being: ligand-gated ion channels, voltage- gated ion channels, other ion channels, nuclear hormone receptors, catalytic receptors, enzymes and transporters. These are presented with nomenclature guidance and summary information on the best available pharmacological tools, alongside key references and suggestions for further reading. The Concise Guide is published in landscape format in order to facilitate comparison of related targets. -

Retrograde Inhibition by a Specific Subset of Interpeduncular Α5 Nicotinic Neurons Regulates Nicotine Preference
Retrograde inhibition by a specific subset of interpeduncular α5 nicotinic neurons regulates nicotine preference Jessica L. Ablesa,b,c, Andreas Görlicha,1, Beatriz Antolin-Fontesa,2,CuidongWanga, Sylvia M. Lipforda, Michael H. Riada, Jing Rend,e,3,FeiHud,e,4,MinminLuod,e,PaulJ.Kennyc, Nathaniel Heintza,f,5, and Ines Ibañez-Tallona,5 aLaboratory of Molecular Biology, The Rockefeller University, New York, NY 10065; bDepartment of Psychiatry, Icahn School of Medicine at Mount Sinai, New York, NY 10029; cDepartment of Neuroscience, Icahn School of Medicine at Mount Sinai, New York, NY 10029; dNational Institute of Biological Sciences, Beijing 102206, China; eSchool of Life Sciences, Tsinghua University, Beijing 100084, China; and fHoward Hughes Medical Institute, The Rockefeller University, New York, NY 10065 Contributed by Nathaniel Heintz, October 23, 2017 (sent for review October 5, 2017; reviewed by Jean-Pierre Changeux and Lorna W. Role) Repeated exposure to drugs of abuse can produce adaptive changes nicotine withdrawal, and optical activation of IPN GABAergic cells that lead to the establishment of dependence. It has been shown that is sufficient to produce a withdrawal syndrome, while blockade of allelic variation in the α5 nicotinic acetylcholine receptor (nAChR) gene GABAergic cells in the IPN reduced symptoms of withdrawal (17). CHRNA5 is associated with higher risk of tobacco dependence. In the Taken together these studies highlight the critical role of α5in brain, α5-containing nAChRs are expressed at very high levels in the regulating behavioral responses to nicotine. Here we characterize two subpopulations of GABAergic interpeduncular nucleus (IPN). Here we identified two nonoverlapping Amigo1 Epyc α + α Amigo1 α Epyc neurons in the IPN that express α5: α5- and α5- neu- 5 cell populations ( 5- and 5- ) in mouse IPN that respond α Amigo1 α Epyc differentially to nicotine. -

Retrograde Inhibition by a Specific Subset of Interpeduncular Α5 Nicotinic Neurons Regulates Nicotine Preference
Retrograde inhibition by a specific subset of interpeduncular α5 nicotinic neurons regulates nicotine preference Jessica L. Ablesa,b,c, Andreas Görlicha,1, Beatriz Antolin-Fontesa,2,CuidongWanga, Sylvia M. Lipforda, Michael H. Riada, Jing Rend,e,3,FeiHud,e,4,MinminLuod,e,PaulJ.Kennyc, Nathaniel Heintza,f,5, and Ines Ibañez-Tallona,5 aLaboratory of Molecular Biology, The Rockefeller University, New York, NY 10065; bDepartment of Psychiatry, Icahn School of Medicine at Mount Sinai, New York, NY 10029; cDepartment of Neuroscience, Icahn School of Medicine at Mount Sinai, New York, NY 10029; dNational Institute of Biological Sciences, Beijing 102206, China; eSchool of Life Sciences, Tsinghua University, Beijing 100084, China; and fHoward Hughes Medical Institute, The Rockefeller University, New York, NY 10065 Contributed by Nathaniel Heintz, October 23, 2017 (sent for review October 5, 2017; reviewed by Jean-Pierre Changeux and Lorna W. Role) Repeated exposure to drugs of abuse can produce adaptive changes nicotine withdrawal, and optical activation of IPN GABAergic cells that lead to the establishment of dependence. It has been shown that is sufficient to produce a withdrawal syndrome, while blockade of allelic variation in the α5 nicotinic acetylcholine receptor (nAChR) gene GABAergic cells in the IPN reduced symptoms of withdrawal (17). CHRNA5 is associated with higher risk of tobacco dependence. In the Taken together these studies highlight the critical role of α5in brain, α5-containing nAChRs are expressed at very high levels in the regulating behavioral responses to nicotine. Here we characterize two subpopulations of GABAergic interpeduncular nucleus (IPN). Here we identified two nonoverlapping Amigo1 Epyc α + α Amigo1 α Epyc neurons in the IPN that express α5: α5- and α5- neu- 5 cell populations ( 5- and 5- ) in mouse IPN that respond α Amigo1 α Epyc differentially to nicotine. -

Genetic Analysis of Benign Familial Epilepsies in the First Year of Life in A
Journal of Human Genetics (2018) 63:9–18 https://doi.org/10.1038/s10038-017-0359-x ARTICLE Genetic analysis of benign familial epilepsies in the first year of life in a Chinese cohort 1 1 1 1 1 1 1 1 2 Qi Zeng ● Xiaoling Yang ● Jing Zhang ● Aijie Liu ● Zhixian Yang ● Xiaoyan Liu ● Ye Wu ● Xiru Wu ● Liping Wei ● Yuehua Zhang1 Received: 11 June 2017 / Revised: 9 August 2017 / Accepted: 9 August 2017 / Published online: 13 November 2017 © The Author(s) 2018. This article is published with open access Abstract Benign familial epilepsies that present themselves in the first year of life include benign familial neonatal epilepsy (BFNE), benign familial neonatal-infantile epilepsy (BFNIE) and benign familial infantile epilepsy (BFIE). We used Sanger sequencing and targeted next-generation sequencing to detect gene mutations in a Chinese cohort of patients with these three disorders. A total of 79 families were collected, including 4 BFNE, 7 BFNIE, and 68 BFIE. Genetic testing led to the identification of gene mutations in 60 families (60 out of 79, 75.9%). A total of 42 families had PRRT2 mutations, 9 had KCNQ2 mutations, 8 had SCN2A mutations, and 1 had a GABRA6 mutation. In total three of four BFNE families were KCNQ2 KCNQ2 SCN2A 1234567890();,: 1234567890();,: detected with mutations. Mutations were detected in all BFNIE families, including 3 mutations, 3 mutations, and 1 PRRT2 mutation. Gene mutations were identified in 50 out of 68 BFIE families (73.5%), including 41 PRRT2 mutations (41 out of 68, 60.3%), 5 SCN2A mutations, 3 KCNQ2 mutations, and 1 GABRA6 mutation. -
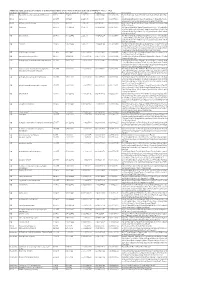
Additional Tables.Xlsx
Additional Table 2 Enriched pathways of downregulated DEGs by the phosphorylation deficient of CRMP2 (P <0.05, q <0.2) Database Description Ratio of DEGs Ratio of universe P -value P adjust q -value Gene name KEGG Biosynthesis of unsaturated fatty acids 8/175 21/4117 1.15E-06 0.000236049 0.000232715 Elovl6/Hsd17b4/Fads2/Elovl4/Scd1/Fads1/Hsd17b12/Hac d3 KEGG Lysosome 15/175 98/4117 1.22E-05 0.00089044 0.000877866 Gla/Man2b1/Entpd4/Gm2a/Acp2/Entpd4/Entpd4b/Scarb 2/Lamp2/Hexa/Asah1/Gns/Lgmn/Smpd1/Ctsd/Ctsb KEGG Fatty acid metabolism 10/175 45/4117 1.30E-05 0.00089044 0.000877866 Elovl6/Acsl1/Hsd17b4/Acaa2/Fads2/Elovl4/Scd1/Fads1/Hs d17b12/Hacd3 GO lysosome 29/455 304/11442 1.14E-05 0.002429124 0.002071406 Gla/Rptor/Man2b1/Gm2a/Acp2/Psen1/Slc35f6/Scarb2/Ba ce1/Snx14/Rnaset2a/Ncstn/Lamp2/Hexa/Plbd2/Asah1/Gn s/Ostm1/Ank3/Pon2/Mlc1/Gja1/Lgmn/Smpd1/Rnf13/Arl8 b/Ctsd/Ctsb/Rab7 GO lytic vacuole 29/455 305/11442 1.21E-05 0.002429124 0.002071406 Gla/Rptor/Man2b1/Gm2a/Acp2/Psen1/Slc35f6/Scarb2/Ba ce1/Snx14/Rnaset2a/Ncstn/Lamp2/Hexa/Plbd2/Asah1/Gn s/Ostm1/Ank3/Pon2/Mlc1/Gja1/Lgmn/Smpd1/Rnf13/Arl8 b/Ctsd/Ctsb/Rab7 GO vacuole 33/455 374/11442 1.47E-05 0.002429124 0.002071406 Gla/Rptor/Abcb6/Man2b1/Entpd4/Gm2a/Acp2/Psen1/Ent pd4b/Slc35f6/Scarb2/Bace1/Snx14/Rnaset2a/Ncstn/Lamp 2/Hexa/Plbd2/Asah1/Gns/Ostm1/Ank3/Pon2/Atg12/Mlc1 /Gja1/Lgmn/Smpd1/Rnf13/Arl8b/Ctsd/Ctsb/Rab7 GO GABA-ergic synapse 12/455 88/11442 0.000178214 0.020677491 0.017632481 Gabrb1/Slitrk3/Iqsec3/Gabrb2/Nlgn3/Gucy1a1/Rims2/Cd h13/Clstn3/Sv2a/Pak1/Atp2b1 GO neuromuscular junction 11/455