Early Response of Methanogenic Archaea to H2 As Evaluated By
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Analyzing Metagenome Data Obtained by High-Throughput Sequencing
Centrum für Biotechnologie Analyzing Metagenome Data Obtained by High-Throughput Sequencing A. Pühler Center for Biotechnology Bielefeld University International Conference: Getting Post 2010 Biodiversity Targets Right Bragança Paulista/SP, Brazil December 11th – 15th, 2010 Content of Talk • Sequence analysis of the metagenome of a model microbial community • Analysis of assembled contigs and single reads by the help of completely sequenced genomes • The functional and taxonomic analysis of single reads using the software programs MetaSAMS and CARMA • The taxonomic analysis of a model microbial community based on 16S-rDNA sequences Sequence Analysis of the Metagenome of a Model Microbial Community (Part I) • Sequencing devices at the CeBiTec of Bielefeld University • Introduction of the model microbial community residing in an agrigultural biogas production • Sequence analysis of the metagenome of the model microbial community High-Throughput Sequencing Devices at the CeBiTec of Bielefeld University Sequencing techniques high-throughput sequencing ABI 3730xl DNA Genome Sequencer Genome Analyzer Analyzer (Applied GS FLX (Roche) (Illumina, Inc.) Biosystems) Genomics Platform Bioinformatics expertise and environment professional data evaluation Bioinformatics Platform Comparison of Different Sequencing Technologies Sequencing techniques ABI 3730xl DNA Genome Sequencer Genome Analyzer Analyzer (Applied GS FLX (Roche) (Illumina, Inc.) Biosystems) read length: 1100 bp 400 bp 150 bp sequenced bases/run: 0,1 Mb 500 Mb 45 Gb The GS FLX system is evidently best suited for a metagenome analysis since it offers long read length combined with an acceptable output. Metagenome Analysis of a Model Microbial Community Residing in a Biogas Production Plant Using Ultrafast Sequencing Biogas production from primary renewable products Biogas is produced during anaerobic digestion of biomass by specific microbial consortia Characteristics of the Analyzed Biogas Plant Located Close to the City of Bielefeld . -

Methanogens Diversity During Anaerobic Sewage Sludge Stabilization and the Effect of Temperature
processes Article Methanogens Diversity during Anaerobic Sewage Sludge Stabilization and the Effect of Temperature Tomáš Vítˇez 1,2, David Novák 3, Jan Lochman 3,* and Monika Vítˇezová 1,* 1 Department of Experimental Biology, Faculty of Science, Masaryk University, 62500 Brno, Czech Republic; [email protected] 2 Department of Agricultural, Food and Environmental Engineering, Faculty of AgriSciences, Mendel University, 61300 Brno, Czech Republic 3 Department of Biochemistry, Faculty of Science, Masaryk University, 62500 Brno, Czech Republic; [email protected] * Correspondence: [email protected] (J.L.); [email protected] (M.V.); Tel.: +420-549-495-602 (J.L.); Tel.: +420-549-497-177 (M.V.) Received: 29 June 2020; Accepted: 10 July 2020; Published: 12 July 2020 Abstract: Anaerobic sludge stabilization is a commonly used technology. Most fermenters are operated at a mesophilic temperature regime. Modern trends in waste management aim to minimize waste generation. One of the strategies can be achieved by anaerobically stabilizing the sludge by raising the temperature. Higher temperatures will allow faster decomposition of organic matter, shortening the retention time, and increasing biogas production. This work is focused on the description of changes in the community of methanogenic microorganisms at different temperatures during the sludge stabilization. At higher temperatures, biogas contained a higher percentage of methane, however, there was an undesirable accumulation of ammonia in the fermenter. Representatives of the hydrogenotrophic genus Methanoliea were described at all temperatures tested. At temperatures up to 50 ◦C, a significant proportion of methanogens were also formed by acetoclastic representatives of Methanosaeta sp. and acetoclastic representatives of the order Methanosarcinales. -

Taxonomic Binning Approaches and Functional Characteristics of the Microbial Community During the Anaerobic Digestion of Hydrolyzed Corncob
energies Article Taxonomic Binning Approaches and Functional Characteristics of the Microbial Community during the Anaerobic Digestion of Hydrolyzed Corncob Luz Breton-Deval 1 , Ilse Salinas-Peralta 1 , Jaime Santiago Alarcón Aguirre 2, Belkis Sulbarán-Rangel 2,* and Kelly Joel Gurubel Tun 2,* 1 Catedras Conacyt, Instituto de Biotecnología, Universidad Nacional Autónoma de México, Cuernavaca 62210, Mexico; [email protected] (L.B.-D.); [email protected] (I.S.-P.) 2 Department of Water and Energy, University of Guadalajara Campus Tonalá, Tonalá 45425, Mexico; [email protected] * Correspondence: [email protected] (B.S.-R.); [email protected] (K.J.G.T.); Tel.: +52-33-2000-2300 (K.J.G.T.) Abstract: Maize forms the basis of Mexican food. As a result, approximately six million tons of corncob are produced each year, which represents an environmental issue, as well as a potential feedstock for biogas production. This research aimed to analyze the taxonomic and functional shift in the microbiome of the fermenters using a whole metagenome shotgun approach. Two strategies were used to understand the microbial community at the beginning and the end of anaerobic digestion: (i) phylogenetic analysis to infer the presence and coverage of clade-specific markers to assign taxonomy and (ii) the recovery of the individual genomes from the samples using the binning of the assembled scaffolds. The results showed that anaerobic digestion brought some noticeable changes and the main microbial community was composed of Corynebacterium variable, Desulfovibrio desulfuricans, Vibrio furnissii, Shewanella spp., Actinoplanes spp., Pseudoxanthomonas spp., Saccharomonospora azurea, Citation: Breton-Deval, L.; Agromyces spp., Serinicoccus spp., Cellulomonas spp., Pseudonocardia spp., Rhodococcus rhodochrous, Salinas-Peralta, I.; Alarcón Aguirre, Sphingobacterium spp. -

Lists of Names of Prokaryotic Candidatus Taxa
NOTIFICATION LIST: CANDIDATUS LIST NO. 1 Oren et al., Int. J. Syst. Evol. Microbiol. DOI 10.1099/ijsem.0.003789 Lists of names of prokaryotic Candidatus taxa Aharon Oren1,*, George M. Garrity2,3, Charles T. Parker3, Maria Chuvochina4 and Martha E. Trujillo5 Abstract We here present annotated lists of names of Candidatus taxa of prokaryotes with ranks between subspecies and class, pro- posed between the mid- 1990s, when the provisional status of Candidatus taxa was first established, and the end of 2018. Where necessary, corrected names are proposed that comply with the current provisions of the International Code of Nomenclature of Prokaryotes and its Orthography appendix. These lists, as well as updated lists of newly published names of Candidatus taxa with additions and corrections to the current lists to be published periodically in the International Journal of Systematic and Evo- lutionary Microbiology, may serve as the basis for the valid publication of the Candidatus names if and when the current propos- als to expand the type material for naming of prokaryotes to also include gene sequences of yet-uncultivated taxa is accepted by the International Committee on Systematics of Prokaryotes. Introduction of the category called Candidatus was first pro- morphology, basis of assignment as Candidatus, habitat, posed by Murray and Schleifer in 1994 [1]. The provisional metabolism and more. However, no such lists have yet been status Candidatus was intended for putative taxa of any rank published in the journal. that could not be described in sufficient details to warrant Currently, the nomenclature of Candidatus taxa is not covered establishment of a novel taxon, usually because of the absence by the rules of the Prokaryotic Code. -

Taxonomy and Ecology of Methanogens
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Horizon / Pleins textes FEMS Microbiology Reviews 87 (1990) 297-308 297 Pubfished by Elsevier FEMSRE 00180 Taxonomy and ecology of methanogens J.L. Garcia Laboratoire de Microbiologie ORSTOM, Université de Provence, Marseille, France Key words: Methanogens; Archaebacteria; Taxonomy; Ecology 1. INTRODUCTION methane from CO2 using alcohols as hydrogen donors; 2-propanol is oxidized to acetone, and More fhan nine reviews on taxonomy of 2-butanol to 2-butanone. Carbon monoxide may methanogens have been published during the last also be converted into methane; most hydro- decade [l-91, after the discovery of the unique genotrophic species (60%) will also use formate. biochemical and genetic properties of these Some aceticlastic species are incapable of oxidiz- organisms led to the concept of Archaebacteria at ing H,. The aceticlastic species of the genus the end of the seventies. Moreover, important Methanosurcina are the most metabolically diverse economic factors have ,placed these bacteria in the methanogens, whereas the obligate aceticlastic limelight [5], including the need to develop alter- Methanosaeta (Methanothrix) can use only acetate. native forms of energy, xenobiotic pollution con- The taxonomy of the methanogenic bacteria trol, the enhancement of meat yields in the cattle has been extensively revised in the light of new industry, the distinction between biological and information based on comparative studies of 16 S thermocatalytic petroleum generation, and the rRNA oligonucleotide sequences, membrane lipid global distribution of methane in the earth's atmo- composition, and antigenic fingerprinting data. sphere. The phenotypic characteristics often do not pro- vide a sufficient means of distinguishing among taxa or determining the phylogenetic position of a 2. -

Microbiology of Lonar Lake and Other Soda Lakes
The ISME Journal (2013) 7, 468–476 & 2013 International Society for Microbial Ecology All rights reserved 1751-7362/13 www.nature.com/ismej MINI REVIEW Microbiology of Lonar Lake and other soda lakes Chakkiath Paul Antony1, Deepak Kumaresan2, Sindy Hunger3, Harold L Drake3, J Colin Murrell4 and Yogesh S Shouche1 1Microbial Culture Collection, National Centre for Cell Science, Pune, India; 2CSIRO Marine and Atmospheric Research, Hobart, TAS, Australia; 3Department of Ecological Microbiology, University of Bayreuth, Bayreuth, Germany and 4School of Environmental Sciences, University of East Anglia, Norwich, UK Soda lakes are saline and alkaline ecosystems that are believed to have existed throughout the geological record of Earth. They are widely distributed across the globe, but are highly abundant in terrestrial biomes such as deserts and steppes and in geologically interesting regions such as the East African Rift valley. The unusual geochemistry of these lakes supports the growth of an impressive array of microorganisms that are of ecological and economic importance. Haloalk- aliphilic Bacteria and Archaea belonging to all major trophic groups have been described from many soda lakes, including lakes with exceptionally high levels of heavy metals. Lonar Lake is a soda lake that is centered at an unusual meteorite impact structure in the Deccan basalts in India and its key physicochemical and microbiological characteristics are highlighted in this article. The occurrence of diverse functional groups of microbes, such as methanogens, methanotrophs, phototrophs, denitrifiers, sulfur oxidizers, sulfate reducers and syntrophs in soda lakes, suggests that these habitats harbor complex microbial food webs that (a) interconnect various biological cycles via redox coupling and (b) impact on the production and consumption of greenhouse gases. -
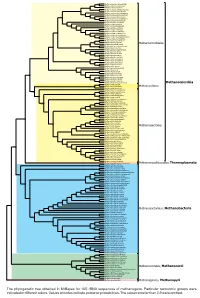
Display Taxonomy in the Phylogenetic Tree
0.90 Methanocalculus natronophilus Methanocalculus taiwanensis 1 Methanocalculus pumilus Methanocalculus chunghsingensis 1 Methanocalculus halotolerans Methanocorpusculum labreanum 1 Methanocorpusculum bavaricum 0.90 Methanocorpusculum sinense Methanocorpusculum aggregans Methanocorpusculum parvum Methanoculleus receptaculi 1 Methanoculleus palmolei Methanoculleus bourgensis 0.93 Methanoculleus sediminis 0.96 0.99 Methanoculleus marisnigri 1 Methanoculleus submarinus Methanoculleus chikugoensis Methanoculleus horonobensis Methanoculleus hydrogenitrophicus 1 Methanoculleus thermophilus 1 Methanoplanus petrolearius 0.95 Methanolacinia paynteri Methanomicrobiales 1 Methanomicrobium mobile Methanoplanus endosymbiosus 1 Methanoplanus limicola 1 Methanogenium organophilum 1 Methanogenium cariaci Methanogenium frigidum Methanogenium boonei 1 Methanogenium marinum 1 Methanofollis formosanus Methanofollis aquaemaris 1 Methanofollis ethanolicus 1 Methanofollis liminatans 1 Methanofollis tationis Methanosphaerula palustris Methanolinea tarda 1 1 Methanoregula boonei 1 Methanoregula formicica 1 1 Methanolinea mesophila 1 Methanospirillum hungatei Methanospirillum stamsii 1 Methanospirillum lacunae 1 Methanospirillum psychrodurum Methanomicrobia 1 Methanocella conradii 1 Methanocella paludicola Methanocellales Methanocella arvoryzae Methermicoccus shengliensis 1 0.98 Methanosaeta harundinacea 1 Methanosaeta pelagica Methanosaeta thermophila 1 1 Methanosaeta concilii 1 Methanohalobium evestigatum Methanosalsum zhilinae 0.97 Methanohalophilus halophilus -

Methanoculleus Marisnigri and Methanogenium, and Description of New Strains of Methanoculleus Bourgense and Met H a Noc U 11 E Us Ma Ris N Igri GLORIA M
INTERNATIONALJOURNAL OF SYSTEMATICBACTERIOLOGY , Apr. 1990, p. 117-122 Vol. 40, No. 2 OO20-7713/90/020117-06$02.OO/O Copyright 0 1990, International Union of Microbiological Societies Transfer of Methanogenium bourgense, Methanogenium marisnigri, Methanogenium olentangyi, and Methanogenium thermophilicum to the Genus Methanoculleus gen. nov., Emendation of Methanoculleus marisnigri and Methanogenium, and Description of New Strains of Methanoculleus bourgense and Met h a noc u 11 e us ma ris n igri GLORIA M. MAESTROJUAN,l DAVID R. BOONE,l* LUYING XUN,2t ROBERT A. MAH,, AND LANFANG ZHANG' Department of Environmental Science and Engineering, Oregon Graduate Center, Beaverton, Oregon 97006-1999, and School of Public Health, University of California, Los Angeles, California 9O01ij2 Two strains of Methanogenium bourgense, strains MS2T(T = type strain) and LX1, were characterized, and, based in part on previously published DNA hybridization data, this species was transferred to a new genus, Methanoculleus, as Methanoculleus bourgense comb. nov. Methanogenium marisnigri JRIT and a new strain of Methanogenium marisnigri, strain ANS, were also characterized. This species was also transferred to the genus Methanoculleus as MethanocuUeus marisnigri comb. nov. et emend., and its description was emended to indicate that the species has a temperature optimum near 40°C, is halotolerant, and is slightly alkaliphilic; in contrast, the previous description of this organism indicates that it has a temperature optimum of 20 to 25"C, is halophilic, and is slightly acidophilic. We also propose the transfer of two other phylogenetically related species, Methanogenium thermophilicum and Methanogenium olentangyi, to the genus Methanoculleus as Methanoculleus thermophilicum and Methanoculleus olentangyi, respectively. Methanogenium cariaci JRIT was also further characterized, and its description is emended. -

Application for Approval to Import Into Containment Any New Organism That
ER-AN-02N 10/02 Application for approval to import into FORM 2N containment any new organism that is not genetically modified, under Section 40 of the Page 1 Hazardous Substances and New Organisms Act 1996 FORM NO2N Application for approval to IMPORT INTO CONTAINMENT ANY NEW ORGANISM THAT IS NOT GENETICALLY MODIFIED under section 40 of the Hazardous Substances and New Organisms Act 1996 Application Title: Importation of extremophilic microorganisms from geothermal sites for research purposes Applicant Organisation: Institute of Geological & Nuclear Sciences ERMA Office use only Application Code: Formally received:____/____/____ ERMA NZ Contact: Initial Fee Paid: $ Application Status: ER-AN-02N 10/02 Application for approval to import into FORM 2N containment any new organism that is not genetically modified, under Section 40 of the Page 2 Hazardous Substances and New Organisms Act 1996 IMPORTANT 1. An associated User Guide is available for this form. You should read the User Guide before completing this form. If you need further guidance in completing this form please contact ERMA New Zealand. 2. This application form covers importation into containment of any new organism that is not genetically modified, under section 40 of the Act. 3. If you are making an application to import into containment a genetically modified organism you should complete Form NO2G, instead of this form (Form NO2N). 4. This form, together with form NO2G, replaces all previous versions of Form 2. Older versions should not now be used. You should periodically check with ERMA New Zealand or on the ERMA New Zealand web site for new versions of this form. -

Methanogenic Community Composition in An
Int. Agrophys., 2017, 31, 327-338 doi: 10.1515/intag-2016-0057 Methanogenic community composition in an organic waste mixture in an anaerobic bioreactor** Agata Gryta1, Karolina Oszust1, Małgorzata Brzezińska1, Krzysztof Ziemiński2, Nina Bilińska-Wielgus1, and Magdalena Frąc1* 1Institute of Agrophysics, Polish Academy of Sciences, Doświadczalna 4, 20-290 Lublin, Poland 2Institute of Fermentation Technology and Microbiology, Łódź University of Technology, Wólczańska 171/173, 90-924 Łódź, Poland Received October 25, 2016; accepted June 1, 2017 A b s t r a c t. The aim of the study was to elucidate the sub- is microbially mediated engineering with the goals of de- stantial relationship between the compositions of methanogen gradation and stabilisation of organic matter, which results community that assembles in the anaerobic digester mass and in the production of energy-rich composites. This process link it to methane production activity. The results of the meta- involves four sequential steps: hydrolysis, fermentation genomic studies were used to evaluate how the methanogen (acidogenesis), acetogenesis (dehydrogenation) and metha- structure changes during an anaerobic digestion process under various waste retention times (21, 23, 25, 29, 33, 39, 47 and 61 nogenesis (acetoclastic or hydrogenotrophic), all of which days). Phylogenetically coherent populations of methanogens depend on the synergistic interactions of microorganisms were assessed by 16S rRNA gene next-generation sequencing and that form a complex metabolic network. This special pro- terminal restriction fragment length polymorphism fingerprint- cess is catalysed especially by methanogenic Archaea. This ing of a specific molecular marker, the mcrA gene. The results unrepeatable group of microorganisms has the capability to indicated multiple phylogenetically diverse methanogen popu- produce methane from basic substrates, such as CO2, H2, ace- lations associated with the various steps of anaerobic digestion. -

Unraveling the Microbiome of a Thermophilic Biogas Plant By
Maus et al. Biotechnol Biofuels (2016) 9:171 DOI 10.1186/s13068-016-0581-3 Biotechnology for Biofuels RESEARCH Open Access Unraveling the microbiome of a thermophilic biogas plant by metagenome and metatranscriptome analysis complemented by characterization of bacterial and archaeal isolates Irena Maus1, Daniela E. Koeck2, Katharina G. Cibis3, Sarah Hahnke4, Yong S. Kim5, Thomas Langer4, Jana Kreubel3, Marcel Erhard6, Andreas Bremges1,7, Sandra Off5, Yvonne Stolze1, Sebastian Jaenicke8, Alexander Goesmann8, Alexander Sczyrba1,7, Paul Scherer5, Helmut König3, Wolfgang H. Schwarz2, Vladimir V. Zverlov2, Wolfgang Liebl2, Alfred Pühler1, Andreas Schlüter1 and Michael Klocke4* Abstract Background: One of the most promising technologies to sustainably produce energy and to mitigate greenhouse gas emissions from combustion of fossil energy carriers is the anaerobic digestion and biomethanation of organic raw material and waste towards biogas by highly diverse microbial consortia. In this context, the microbial systems ecol- ogy of thermophilic industrial-scale biogas plants is poorly understood. Results: The microbial community structure of an exemplary thermophilic biogas plant was analyzed by a com- prehensive approach comprising the analysis of the microbial metagenome and metatranscriptome comple- mented by the cultivation of hydrolytic and acido-/acetogenic Bacteria as well as methanogenic Archaea. Analysis of metagenome-derived 16S rRNA gene sequences revealed that the bacterial genera Defluviitoga (5.5 %), Halocella (3.5 %), Clostridium -

Toward Understanding the Physiological Determinants of Microbial Competitiveness in Methanogenic Processes
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Doctoral Dissertations Graduate School 12-2014 Toward Understanding the Physiological Determinants of Microbial Competitiveness in Methanogenic Processes Si Chen University of Tennessee - Knoxville, [email protected] Follow this and additional works at: https://trace.tennessee.edu/utk_graddiss Part of the Environmental Engineering Commons Recommended Citation Chen, Si, "Toward Understanding the Physiological Determinants of Microbial Competitiveness in Methanogenic Processes. " PhD diss., University of Tennessee, 2014. https://trace.tennessee.edu/utk_graddiss/3191 This Dissertation is brought to you for free and open access by the Graduate School at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Doctoral Dissertations by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. To the Graduate Council: I am submitting herewith a dissertation written by Si Chen entitled "Toward Understanding the Physiological Determinants of Microbial Competitiveness in Methanogenic Processes." I have examined the final electronic copy of this dissertation for form and content and recommend that it be accepted in partial fulfillment of the equirr ements for the degree of Doctor of Philosophy, with a major in Civil Engineering. Qiang He, Major Professor We have read this dissertation and recommend its acceptance: Chris Cox, Terry Hazen, Gary Sayler Accepted for the Council: Carolyn R. Hodges Vice Provost and Dean of the Graduate School (Original signatures are on file with official studentecor r ds.) Toward Understanding the Physiological Determinants of Microbial Competitiveness in Methanogenic Processes A Dissertation Presented for the Doctor of Philosophy Degree The University of Tennessee, Knoxville Si Chen December 2014 Copyright © 2014 by Si Chen All rights reserved ii DEDICATION This dissertation is dedicated in loving memory to my maternal grandfather.