(EVG/COBI/FTC/TAF [E/C/F/TAF] FDC) Gilead Sciences
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
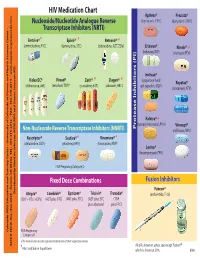
HIV Medication Chart Aptivus® Prezista® Nucleoside/Nucleotide Analogue Reverse (Tipranavir, TPV) (Darunavir, DRV) Transcriptase Inhibitors (NRTI)
HIV Medication Chart Aptivus® Prezista® Nucleoside/Nucleotide Analogue Reverse (tipranavir, TPV) (darunavir, DRV) Transcriptase Inhibitors (NRTI) Emtriva®* Epivir® * Retrovir® * (emtricitabine, FTC) (lamivudine, 3TC) (zidovudine, AZT, ZDV) Crixivan® Norvir® * (indinavir, IDV) (ritonavir, RTV) Invirase® Videx EC® Viread® Zerit® Ziagen® * * (saquinavir hard Reyataz® (didanosine, ddl) (tenofovir,TDF)* (stavudine, d4T) (abacavir, ABC) gel capsules, SQV) (atazanavir, ATV) Kaletra® * (lopinavir/ritonavir, LPV/r) Viracept® Non-Nucleoside Reverse Transcriptase Inhibitors (NNRTI) Inhibitors (PI) Protease (nelfinavir, NFV) Rescriptor® Sustiva®* Viramune® * (delavirdine, DLV) (efavirenz, EFV) (nevirapine, NVP) Lexiva® (fosamprenavir, FPV) FDA Pregnancy Category D Fixed Dose Combinations Fusion Inhibitors Fuzeon® Developed by MeriLou Johnson, MeriLou by Developed Johnson, MSW, and Steven MPA MD Atripla® Combivir® Epzicom® Trizivir® Truvada® (enfuvirtide,T-20) (TDF+FTC+EFV) (AZT plus 3TC) (ABC plus 3TC) (AZT plus 3TC (TDF plus abacavir) plus FTC) Colorado AIDS Education and Training Center • University of Colorado at Denver and Health Sciences Center • Center and Health Sciences Denver at of Colorado • University Center Training and AIDS Education Colorado FDA Pregnancy Category D ®The brands listed are the registered trademarks of their respective owners. 4200 E. Ave., Ninth TEL: A089 • Denver, Box 80262 CO 303-315-2512 • FAX: • 303-315-2514 • www.mpaetc.org/colorado.htm All pills shown in actual size except Fuzeon® *Also available in liquid form. which is shown at 50%. 8/06 Medication Schedule Name______________________________ Date___________ Number of Time of day you ar to take TOTAL Name of Medication pills to take this medicine Food Interactions Side Effects number of each time pills each day With Food Without Food ❑ ❑ With Food Without Food ❑ ❑ With Food Without Food ❑ ❑ With Food Without Food ❑ ❑ With Food Without Food ❑ ❑ With Food Without Food ❑ ❑ Discontinued Medications or Formulations Helpful Hints: • Refill prescriptions before you run out. -

Truvada (Emtricitabine / Tenofovir Disoproxil)
Pre-exposure Prophylaxis (2.3) HIGHLIGHTS OF PRESCRIBING INFORMATION These highlights do not include all the information needed to use Recommended dose in HIV-1 uninfected adults: One tablet TRUVADA safely and effectively. See full prescribing information (containing 200 mg/300 mg of emtricitabine and tenofovir for TRUVADA. disoproxil fumarate) once daily taken orally with or without food. (2.3) TRUVADA® (emtricitabine/tenofovir disoproxil fumarate) tablets, for oral use Recommended dose in renally impaired HIV-uninfected Initial U.S. Approval: 2004 individuals: Do not use TRUVADA in HIV-uninfected individuals if CrCl is below 60 mL/min. If a decrease in CrCl is observed in WARNING: LACTIC ACIDOSIS/SEVERE HEPATOMEGALY WITH uninfected individuals while using TRUVADA for PrEP, evaluate STEATOSIS, POST-TREATMENT ACUTE EXACERBATION OF potential causes and re-assess potential risks and benefits of HEPATITIS B, and RISK OF DRUG RESISTANCE WITH USE OF continued use. (2.4) TRUVADA FOR PrEP IN UNDIAGNOSED HIV-1 INFECTION -----------------------DOSAGE FORMS AND STRENGTHS------------------- See full prescribing information for complete boxed warning. Tablets: 200 mg/300 mg, 167 mg/250 mg, 133 mg/200 mg, and 100 Lactic acidosis and severe hepatomegaly with steatosis, mg/150 mg of emtricitabine and tenofovir disoproxil fumarate . (3) including fatal cases, have been reported with the use of nucleoside analogs, including VIREAD, a component of TRUVADA. (5.1) --------------------------------CONTRAINDICATIONS----------------------------- TRUVADA is not approved for the treatment of chronic Do not use TRUVADA for pre-exposure prophylaxis in individuals with hepatitis B virus (HBV) infection. Severe acute unknown or positive HIV-1 status. TRUVADA should be used in exacerbations of hepatitis B have been reported in patients HIV-infected patients only in combination with other antiretroviral coinfected with HIV-1 and HBV who have discontinued agents. -

Page: Treatment-Drugs
© National HIV Curriculum PDF created September 29, 2021, 5:12 am Darunavir-Cobicistat-Tenofovir alafenamide-Emtricitabine (Symtuza) Table of Contents Darunavir-Cobicistat-Tenofovir alafenamide-Emtricitabine Symtuza Summary Drug Summary Key Clinical Trials Key Drug Interactions Drug Summary The fixed-dose combination tablet darunavir-cobicistat-tenofovir alafenamide-emtricitabine is a single-tablet regimen that can be considered for treatment-naïve or certain treatment-experienced adults living with HIV. This single-tablet regimen offers a one pill daily regimen with high barrier to resistance (due to the darunavir- cobicistat), with potentially less renal and bone toxicity as compared to regimens that include tenofovir DF; however, it has potential gastrointestinal adverse effects and drug-drug interactions, primarily due to the cobicistat component. In clinical trials, darunavir-cobicistat-tenofovir alafenamide-emtricitabine was compared to darunavir-cobicistat plus tenofovir DF-emtricitabine as initial therapy for treatment-naïve individuals and found to be equally effective in terms of viral suppression. A switch to the fixed-dose combination tablet was also compared to continuing a boosted protease inhibitor plus tenofovir DF- emtricitabine and again determined to have equivalent efficacy. The FDA has approved darunavir-cobicistat- tenofovir alafenamide-emtricitabine as a complete regimen for treatment-naïve individuals or treatment- experienced individuals who have a suppressed HIV RNA level on a stable regimen for at least 6 months and no resistance to darunavir or tenofovir. Key Clinical Trials A phase 3 trial in treatment-naïve individuals compared the fixed-dose single-tablet regimen darunavir- cobicistat-tenofovir alafenamide-emtricitabine with the regimen darunavir-cobicistat plus tenofovir DF- emtricitabine emtricitabine [AMBER]. -

Download Article PDF/Slides
New Antiretrovirals in Development: Reprinted from The PRN Notebook,™ june 2002. Dr. James F. Braun, Editor-in-Chief. Tim Horn, Executive Editor. Published in New York City by the Physicians’ Research Network, Inc.,® John Graham Brown, Executive Director. For further information and other articles The View in 2002 available online, visit http://www.PRN.org All rights reserved. © june 2002. Roy “Trip” Gulick, md, mph Associate Professor of Medicine, Weill Medical College of Cornell University Director, Cornell Clinical Trials Unit, New York, New York Summary by Tim Horn Edited by Scott Hammer, md espite the fact that 16 antiretro- tiviral activity of emtricitabine was estab- Preliminary results from two random- virals are approved for use in the lished, with total daily doses of 200 mg or ized studies—FTC-302 and FTC-303—were United States, there is an indis- more producing the greatest median viral reported by Dr. Charles van der Horst and putable need for new anti-hiv com- load suppression: 1.72-1.92 log. Based on his colleagues at the 8th croi, held in Feb- pounds that have potent and these data, a once-daily dose of 200 mg ruary 2001 in Chicago (van der Horst, durable efficacy profiles, unique re- was selected for further long-term clinical 2001). FTC-302 was a blinded comparison sistance patterns, patient-friendly dosing study. “This is what we’re looking forward of emtricitabine and lamivudine, both in schedules, and minimal toxicities. To pro- to with emtricitabine,” commented Dr. combination with stavudine (Zerit) and vide prn with a glimpse of drugs current- Gulick. -

Product Monograph for CELSENTRI
PRODUCT MONOGRAPH PrCELSENTRI maraviroc Tablets 150 and 300 mg CCR5 antagonist ViiV Healthcare ULC 245, boulevard Armand-Frappier Laval, Quebec H7V 4A7 Date of Revision: July 05, 2019 Submission Control No: 226222 © 2019 ViiV Healthcare group of companies or its licensor. Trademarks are owned by or licensed to the ViiV Healthcare group of companies. Page 1 of 60 Table of Contents PART I: HEALTH PROFESSIONAL INFORMATION.........................................................3 SUMMARY PRODUCT INFORMATION ........................................................................3 INDICATIONS AND CLINICAL USE..............................................................................3 CONTRAINDICATIONS ...................................................................................................3 WARNINGS AND PRECAUTIONS..................................................................................4 ADVERSE REACTIONS....................................................................................................9 DRUG INTERACTIONS ..................................................................................................19 DOSAGE AND ADMINISTRATION..............................................................................28 OVERDOSAGE ................................................................................................................31 ACTION AND CLINICAL PHARMACOLOGY ............................................................31 STORAGE AND STABILITY..........................................................................................36 -

WO 2017/004012 Al 5 January 2017 (05.01.2017) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2017/004012 Al 5 January 2017 (05.01.2017) P O P C T (51) International Patent Classification: AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, A61K 9/24 (2006.01) A61K 31/513 (2006.01) BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, A61K 31/505 (2006.01) A61K 31/675 (2006.01) DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, (21) International Application Number: KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, PCT/US20 16/039762 MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, (22) International Filing Date: PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, 28 June 2016 (28.06.2016) SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (25) Filing Language: English (84) Designated States (unless otherwise indicated, for every (26) Publication Language: English kind of regional protection available): ARIPO (BW, GH, (30) Priority Data: GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, 62/187,102 30 June 2015 (30.06.2015) US TZ, UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, 62/296,524 17 February 2016 (17.02.2016) US TJ, TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, DK, EE, ES, FI, FR, GB, GR, HR, HU, IE, IS, IT, LT, LU, (71) Applicants: GILEAD SCIENCES, INC. -

Tenofovir Alafenamide Rescues Renal Tubules in Patients with Chronic Hepatitis B
life Communication Tenofovir Alafenamide Rescues Renal Tubules in Patients with Chronic Hepatitis B Tomoya Sano * , Takumi Kawaguchi , Tatsuya Ide, Keisuke Amano, Reiichiro Kuwahara, Teruko Arinaga-Hino and Takuji Torimura Division of Gastroenterology, Department of Medicine, Kurume University School of Medicine, Kurume, Fukuoka 830-0011, Japan; [email protected] (T.K.); [email protected] (T.I.); [email protected] (K.A.); [email protected] (R.K.); [email protected] (T.A.-H.); [email protected] (T.T.) * Correspondence: [email protected]; Tel.: +81-942-31-7627 Abstract: Nucles(t)ide analogs (NAs) are effective for chronic hepatitis B (CHB). NAs suppress hepatic decompensation and hepatocarcinogenesis, leading to a dramatic improvement of the natural course of patients with CHB. However, renal dysfunction is becoming an important issue for the management of CHB. Renal dysfunction develops in patients with the long-term treatment of NAs including adefovir dipivoxil and tenofovir disoproxil fumarate. Recently, several studies have reported that the newly approved tenofovir alafenamide (TAF) has a safe profile for the kidney due to greater plasma stability. In this mini-review, we discuss the effectiveness of switching to TAF for NAs-related renal tubular dysfunction in patients with CHB. Keywords: adefovir dipivoxil (ADV); Fanconi syndrome; hepatitis B virus (HBV); renal tubular Citation: Sano, T.; Kawaguchi, T.; dysfunction; tenofovir alafenamide (TAF); tenofovir disoproxil fumarate (TDF); β2-microglobulin Ide, T.; Amano, K.; Kuwahara, R.; Arinaga-Hino, T.; Torimura, T. Tenofovir Alafenamide Rescues Renal Tubules in Patients with Chronic 1. -

Annex I Summary of Product Characteristics
ANNEX I SUMMARY OF PRODUCT CHARACTERISTICS 1 1. NAME OF THE MEDICINAL PRODUCT VIRACEPT 250 mg film-coated tablets 2. QUALITATIVE AND QUANTITATIVE COMPOSITION VIRACEPT 250 mg film-coated tablets contain 292.25 mg of nelfinavir mesylate corresponding to 250 mg of nelfinavir (as free base). For excipients, see 6.1. 3. PHARMACEUTICAL FORM Film-coated tablets 4. CLINICAL PARTICULARS 4.1 Therapeutic indications VIRACEPT is indicated in antiretroviral combination treatment of human immunodeficiency virus (HIV-1) infected adults, adolescents and children of 3 years of age and older. In protease inhibitor experienced patients the choice of nelfinavir should be based on individual viral resistance testing and treatment history. Refer to Section 5.1 Pharmacodynamic properties. 4.2 Posology and method of administration VIRACEPT film-coated tablets are administered orally and should be ingested with food. Patients older than 13 years: the recommended dosage of VIRACEPT film-coated tablets is 1250 mg (five 250 mg tablets) twice a day (BID) or 750 mg (three 250 mg tablets) three times a day (TID) by mouth. The efficacy of the BID regimen has been evaluated versus the TID regimen primarily in patients naïve to protease inhibitors (see section 5.1, pharmacodynamic properties). Patients aged 3 to 13 years: for children, the recommended starting dose is 25 – 30 mg/kg body weight per dose given TID. For children unable to take tablets, VIRACEPT oral powder may be administered (see Summary of Product Characteristics for VIRACEPT oral powder). The recommended dose of VIRACEPT film-coated tablets to be administered TID to children aged 3 to 13 years is as follows: Body Weight Number of Viracept 250mg kg film-coated tablets* 18 to < 23 2 ≥ 23 3 * see Summary of Product Characteristics for VIRACEPT oral powder for patients with less than 18 kg body weight. -

DESCOVY, and Upon Diagnosis of These Highlights Do Not Include All the Information Needed to Use Any Other Sexually Transmitted Infections (Stis)
HIGHLIGHTS OF PRESCRIBING INFORMATION once every 3 months while taking DESCOVY, and upon diagnosis of These highlights do not include all the information needed to use any other sexually transmitted infections (STIs). (2.2) DESCOVY safely and effectively. See full prescribing information • Recommended dosage: for DESCOVY. • Treatment of HIV-1 Infection: One tablet taken once daily with or ® without food in patients with body weight at least 25 kg. (2.3) DESCOVY (emtricitabine and tenofovir alafenamide) tablets, for • HIV-1 PrEP: One tablet taken once daily with or without food in oral use individuals with body weight at least 35 kg. (2.4) Initial U.S. Approval: 2015 • Renal impairment: DESCOVY is not recommended in individuals with WARNING: POST-TREATMENT ACUTE EXACERBATION OF estimated creatinine clearance below 30 mL per minute. (2.5) HEPATITIS B and RISK OF DRUG RESISTANCE WITH USE ----------------------DOSAGE FORMS AND STRENGTHS-------------------- OF DESCOVY FOR HIV-1 PRE-EXPOSURE PROPHYLAXIS Tablets: 200 mg of FTC and 25 mg of TAF (3) (PrEP) IN UNDIAGNOSED EARLY HIV-1 INFECTION See full prescribing information for complete boxed warning. -------------------------------CONTRAINDICATIONS------------------------------ DESCOVY for HIV-1 PrEP is contraindicated in individuals with Severe acute exacerbations of hepatitis B (HBV) have been unknown or positive HIV-1 status. (4) reported in HBV-infected individuals who have discontinued products containing emtricitabine (FTC) and/or tenofovir -----------------------WARNINGS AND PRECAUTIONS----------------------- disoproxil fumarate (TDF), and may occur with • Comprehensive management to reduce the risk of sexually discontinuation of DESCOVY. Hepatic function should be transmitted infections (STIs), including HIV-1, when DESCOVY is monitored closely in these individuals. -

Switching from Efavirenz, Emtricitabine, and Tenofovir Disoproxil Fumarate
Articles Switching from efavirenz, emtricitabine, and tenofovir disoproxil fumarate to tenofovir alafenamide coformulated with rilpivirine and emtricitabine in virally suppressed adults with HIV-1 infection: a randomised, double-blind, multicentre, phase 3b, non-inferiority study Edwin DeJesus, Moti Ramgopal, Gordon Crofoot, Peter Ruane, Anthony LaMarca, Anthony Mills, Claudia T Martorell, Joseph de Wet, Hans-Jürgen Stellbrink, Jean-Michel Molina, Frank A Post, Ignacio Pérez Valero, Danielle Porter, YaPei Liu, Andrew Cheng, Erin Quirk, Devi SenGupta, Huyen Cao Summary Background Tenofovir alafenamide is a prodrug that reduces tenofovir plasma concentrations by 90% compared with Lancet HIV 2017 tenofovir disoproxil fumarate, thereby decreasing bone and renal risks. The coformulation of rilpivirine, emtricitabine, Published Online and tenofovir alafenamide has recently been approved, and we aimed to investigate the efficacy, safety, and tolerability March 1, 2017 of switching to this regimen compared with remaining on coformulated efavirenz, emtricitabine, and tenofovir http://dx.doi.org/10.1016/ S2352-3018(17)30032-2 disoproxil fumarate. See Online/Articles http://dx.doi.org/10.1016/ Methods In this randomised, double-blind, placebo-controlled, non-inferiority trial, HIV-1-infected adults were S2352-3018(17)30031-0 enrolled at 120 hospitals and outpatient clinics in eight countries in North America and Europe. Participants were See Online/Comment virally suppressed (HIV-1 RNA <50 copies per mL) on efavirenz, emtricitabine, and tenofovir -

(12) Patent Application Publication (10) Pub. No.: US 2008/0241135 A1 Olson Et Al
US 20080241135A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2008/0241135 A1 Olson et al. (43) Pub. Date: Oct. 2, 2008 (54) METHODS FOR REDUCINGVIRAL LOAD IN 60/711,528, filed on Aug. 26, 2005, provisional appli HIV-1-INFECTED PATIENTS cation No. 60/715,619, filed on Sep. 9, 2005. (75) Inventors: William C. Olson, Ossining, NY Publication Classification (US); Paul J. Maddon, Scarsdale, NY (US); Daniel C. Pevear, (51) Int. Cl. Medford, MA (US); Robert J. A 6LX 39/395 (2006.01) Israel, Suffern, NY (US); Jose D. A6IP 43/00 (2006.01) Murga, Rosedale, NY (US) (52) U.S. Cl. ..................................................... 424/133.1 Correspondence Address: (57) ABSTRACT COOPER & DUNHAM, LLP 1185 AVENUE OF THE AMERICAS This method provides a method for reducing HIV-1 viral load NEW YORK, NY 10036 in an HIV-1-infected human subject which comprises admin istering to the subject at a predefined interval effective HIV-1 (73) Assignee: Progenics Pharmaceuticals, Inc. viral load-reducing doses of (a) a humanized antibody desig nated PRO 140, or of (b) an anti-CCR5 receptor monoclonal (21) Appl. No.: 11/894,762 antibody. This invention also provides a method for inhibiting in a human Subject the onset or progression of an HIV-1- (22) Filed: Aug. 20, 2007 associated disorder, the inhibition of which is effected by inhibiting fusion of HIV-1 to CCR5"CD4" target cells in the Related U.S. Application Data Subject. This invention also provides a method for treating a subject infected with HIV-1 comprising administering to the (63) Continuation of application No. -

Page: Treatment-Drugs
© National HIV Curriculum PDF created September 27, 2021, 11:42 pm Saquinavir (Invirase) Table of Contents Saquinavir Invirase Summary Drug Summary Key Clinical Trials Resistance Key Drug Interactions Drug Summary Saquinavir, the first protease inhibitor approved by the FDA (in 1995), played a central part in the initial roll out of highly active combination antiretroviral therapy in the mid-1990’s. Three formulations of saquinavir have been approved, including the initial hard-gel capsule, a soft-gel capsule (now discontinued), and, more recently, a tablet. In the early years of combination antiretroviral therapy, unboosted saquinavir was used in combination with two nucleoside reverse transcriptase inhibitors (NRTIs). Subsequently, saquinavir has been used in combination with low-dose ritonavir boosting. Over time, multiple better-tolerated and more convenient antiretroviral agents have become available and have replaced saquinavir. At this time, saquinavir is no longer recommended for use in antiretroviral therapy regimens and patients taking this agent should switch to a currently recommended option. Key Clinical Trials Early trials demonstrated that saquinavir plus two NRTI’s (zidovudine and zalcitabine) led to greater reductions in HIV RNA and increases in CD4 count as compared to dual therapy with zidovudine plus saquinavir or zidovudine plus zalcitabine [ACTG 229]. Subsequently, saquinavir was compared to other protease inhibitors, each given with two NRTIs. One study demonstrated similar antiviral potency between saquinavir boosted with ritonavir and lopinavir-ritonavir, each given with tenofovir DF-emtricitabine [GEMINI], whereas a similar trial demonstrated higher rates of treatment failure and discontinuation in the saquinavir plus ritonavir arm and concluded that lopinavir-ritonavir has superior antiviral efficacy, largely driven by tolerability and patient preference [MaxCmin2].