IDENTIFICATION of CANDIDATE GENES and ISOFORMS ASSOCIATED with GENETIC RESISTANCE to MAREK's DISEASE from RNA-SEQ DATA by Liki
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Genetic and Genomic Analysis of Hyperlipidemia, Obesity and Diabetes Using (C57BL/6J × TALLYHO/Jngj) F2 Mice
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Nutrition Publications and Other Works Nutrition 12-19-2010 Genetic and genomic analysis of hyperlipidemia, obesity and diabetes using (C57BL/6J × TALLYHO/JngJ) F2 mice Taryn P. Stewart Marshall University Hyoung Y. Kim University of Tennessee - Knoxville, [email protected] Arnold M. Saxton University of Tennessee - Knoxville, [email protected] Jung H. Kim Marshall University Follow this and additional works at: https://trace.tennessee.edu/utk_nutrpubs Part of the Animal Sciences Commons, and the Nutrition Commons Recommended Citation BMC Genomics 2010, 11:713 doi:10.1186/1471-2164-11-713 This Article is brought to you for free and open access by the Nutrition at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Nutrition Publications and Other Works by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. Stewart et al. BMC Genomics 2010, 11:713 http://www.biomedcentral.com/1471-2164/11/713 RESEARCH ARTICLE Open Access Genetic and genomic analysis of hyperlipidemia, obesity and diabetes using (C57BL/6J × TALLYHO/JngJ) F2 mice Taryn P Stewart1, Hyoung Yon Kim2, Arnold M Saxton3, Jung Han Kim1* Abstract Background: Type 2 diabetes (T2D) is the most common form of diabetes in humans and is closely associated with dyslipidemia and obesity that magnifies the mortality and morbidity related to T2D. The genetic contribution to human T2D and related metabolic disorders is evident, and mostly follows polygenic inheritance. The TALLYHO/ JngJ (TH) mice are a polygenic model for T2D characterized by obesity, hyperinsulinemia, impaired glucose uptake and tolerance, hyperlipidemia, and hyperglycemia. -

Serum Albumin OS=Homo Sapiens
Protein Name Cluster of Glial fibrillary acidic protein OS=Homo sapiens GN=GFAP PE=1 SV=1 (P14136) Serum albumin OS=Homo sapiens GN=ALB PE=1 SV=2 Cluster of Isoform 3 of Plectin OS=Homo sapiens GN=PLEC (Q15149-3) Cluster of Hemoglobin subunit beta OS=Homo sapiens GN=HBB PE=1 SV=2 (P68871) Vimentin OS=Homo sapiens GN=VIM PE=1 SV=4 Cluster of Tubulin beta-3 chain OS=Homo sapiens GN=TUBB3 PE=1 SV=2 (Q13509) Cluster of Actin, cytoplasmic 1 OS=Homo sapiens GN=ACTB PE=1 SV=1 (P60709) Cluster of Tubulin alpha-1B chain OS=Homo sapiens GN=TUBA1B PE=1 SV=1 (P68363) Cluster of Isoform 2 of Spectrin alpha chain, non-erythrocytic 1 OS=Homo sapiens GN=SPTAN1 (Q13813-2) Hemoglobin subunit alpha OS=Homo sapiens GN=HBA1 PE=1 SV=2 Cluster of Spectrin beta chain, non-erythrocytic 1 OS=Homo sapiens GN=SPTBN1 PE=1 SV=2 (Q01082) Cluster of Pyruvate kinase isozymes M1/M2 OS=Homo sapiens GN=PKM PE=1 SV=4 (P14618) Glyceraldehyde-3-phosphate dehydrogenase OS=Homo sapiens GN=GAPDH PE=1 SV=3 Clathrin heavy chain 1 OS=Homo sapiens GN=CLTC PE=1 SV=5 Filamin-A OS=Homo sapiens GN=FLNA PE=1 SV=4 Cytoplasmic dynein 1 heavy chain 1 OS=Homo sapiens GN=DYNC1H1 PE=1 SV=5 Cluster of ATPase, Na+/K+ transporting, alpha 2 (+) polypeptide OS=Homo sapiens GN=ATP1A2 PE=3 SV=1 (B1AKY9) Fibrinogen beta chain OS=Homo sapiens GN=FGB PE=1 SV=2 Fibrinogen alpha chain OS=Homo sapiens GN=FGA PE=1 SV=2 Dihydropyrimidinase-related protein 2 OS=Homo sapiens GN=DPYSL2 PE=1 SV=1 Cluster of Alpha-actinin-1 OS=Homo sapiens GN=ACTN1 PE=1 SV=2 (P12814) 60 kDa heat shock protein, mitochondrial OS=Homo -

List of Down-Regulated Transcripts
Supplementary Table 1: List of down-regulated transcripts (Fold Change>1.7 and p<0.05). List of the transcipts downregulated by Tpr-Met in P27 cachectic gastrocnemius muscles. Shown are the transcripts gene annotation: Mus Musculus Entrez Gene ID, official symbol from NCBI, average (AVG) Log2 Ratio derived from Illumina microarrays (3 replicates/condition), fold change and p-value computations, Log2 ratio over AVG Log2 controls and full definition of the genes. All genes modulated with a fold change > 1.7 and p-value<0.05 are included. AVG Entrez Gene Fold Symbol Log2 T-test Ctrl Ctrl Ctrl TM TM TM Definition ID Change Ratio - - - - 26908 Eif2s3y 3.598 12.1 0.000 0.146 0.248 0.102 3.712 3.571 -3.510 eukaryotic translation initiation factor 2, subunit 3, structural gene Y-linked (Eif2s3y), mRNA. - - - 13628 Eef1a2 1.333 2.5 0.011 -0.444 0.261 0.183 1.503 0.950 -1.546 eukaryotic translation elongation factor 1 alpha 2 (Eef1a2), mRNA. - - - 13628 Eef1a2 1.298 2.5 0.031 -0.383 0.275 0.108 1.632 0.605 -1.656 eukaryotic translation elongation factor 1 alpha 2 (Eef1a2), mRNA. - - - 56222 Cited4 1.268 2.4 0.029 -0.466 0.035 0.431 1.109 0.889 -1.805 Cbp/p300-interacting transactivator, with Glu/Asp-rich carboxy-terminal domain, 4 (Cited4), mRNA. - - - 18810 Plec1 1.246 2.4 0.022 -0.440 0.327 0.113 1.303 0.779 -1.657 plectin 1 (Plec1), transcript variant 10, mRNA. - - - 20741 Spnb1 1.238 2.4 0.016 -0.384 0.314 0.071 1.129 0.902 -1.684 spectrin beta 1 (Spnb1), mRNA. -
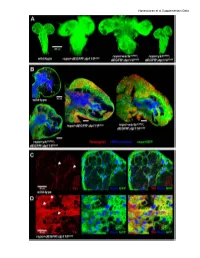
Supplementary Data Vigneswaran Et Al Supplementary Data
Vigneswaran et al Supplementary Data Vigneswaran et al Supplementary Data Figure S1: Yki is required for EGFR-PI3K-driven glial neoplasia in Drosophila (A) Optical projections of whole brain-nerve cord complexes from 3rd instar larvae approximately 130 hrs old. Dorsal view; anterior up. CD8-GFP (green) labels glial cell bodies. Compared to repo>dEGFRλ;dp110CAAX, warts knockdown (repo>wartsdsRNA; dEGFRλ;dp110CAAX) increased neoplastic brain overgrowth and yki knockdown (repo>ykidsRNA;dEGFRλ;dp110CAAX) decreased neoplastic brain overgrowth. (B) 3 µm optical projections of brain hemispheres, age-matched 3rd instar larvae. Frontal sections; anterior up; midline to left. Repo (red) labels glial cell nuclei; CD8-GFP (green) labels glial cell bodies; anti-HRP (blue) counter-stains for neurons and neuropil. (middle) repo>dEGFRλ;dp110CAAX showed increased glial cell numbers (red nuclei) compared to (upper left) wild-type. Compared to repo>dEGFRλ;dp110CAAX, (right) warts knockdown increased neoplastic glial cell numbers (red nuclei), whereas (lower left) yki knockdown reduced neoplastic glial cell numbers (red nuclei). (C, D) Low levels of Yki protein (red) was observed in wild-type central brain glia (white arrows, left panel in C) compared to high levels of cytoplasmic and nuclear Yki protein in dEGFRλ;dp110CAAX neoplastic glia (white arrows, left panel in D); Repo (blue) labels glial cell nuclei; CD8-GFP (green) labels glial cell bodies. Vigneswaran et al Supplementary Data Figure S2: YAP/TAZ expression confined to RTK-amplified tuMor cells and Maintained in patient-derived xenografts (A) On the left, immunohistochemical (IHC) staining in representative normal brain parenchyma in the cortex where YAP expression and TAZ expression was limited to vascular cells and was not detectable in normal neuronal and glial cells. -

SUPPLEMENTARY APPENDIX Inflammation Regulates Long Non-Coding RNA-PTTG1-1:1 in Myeloid Leukemia
SUPPLEMENTARY APPENDIX Inflammation regulates long non-coding RNA-PTTG1-1:1 in myeloid leukemia Sébastien Chateauvieux, 1,2 Anthoula Gaigneaux, 1° Déborah Gérard, 1 Marion Orsini, 1 Franck Morceau, 1 Barbora Orlikova-Boyer, 1,2 Thomas Farge, 3,4 Christian Récher, 3,4,5 Jean-Emmanuel Sarry, 3,4 Mario Dicato 1 and Marc Diederich 2 °Current address: University of Luxembourg, Faculty of Science, Technology and Communication, Life Science Research Unit, Belvaux, Luxemburg. 1Laboratoire de Biologie Moléculaire et Cellulaire du Cancer, Hôpital Kirchberg, Luxembourg, Luxembourg; 2College of Pharmacy, Seoul National University, Gwanak-gu, Seoul, Korea; 3Cancer Research Center of Toulouse, UMR 1037 INSERM/ Université Toulouse III-Paul Sabatier, Toulouse, France; 4Université Toulouse III Paul Sabatier, Toulouse, France and 5Service d’Hématologie, Centre Hospitalier Universitaire de Toulouse, Institut Universitaire du Cancer de Toulouse Oncopôle, Toulouse, France Correspondence: MARC DIEDERICH - [email protected] doi:10.3324/haematol.2019.217281 Supplementary data Inflammation regulates long non-coding RNA-PTTG1-1:1 in myeloid leukemia Sébastien Chateauvieux1,2, Anthoula Gaigneaux1*, Déborah Gérard1, Marion Orsini1, Franck Morceau1, Barbora Orlikova-Boyer1,2, Thomas Farge3,4, Christian Récher3,4,5, Jean-Emmanuel Sarry3,4, Mario Dicato1 and Marc Diederich2 1 Laboratoire de Biologie Moléculaire et Cellulaire du Cancer, Hôpital Kirchberg, 9, rue Edward Steichen, 2540 Luxembourg, Luxemburg; 2 College of Pharmacy, Seoul National University, 1 Gwanak-ro, -

New Partners Identified by Mass Spectrometry Assay Reveal Functions of NCAM2 in Neural Cytoskeleton Organization
International Journal of Molecular Sciences Article New Partners Identified by Mass Spectrometry Assay Reveal Functions of NCAM2 in Neural Cytoskeleton Organization Antoni Parcerisas 1,2,3,*,† , Alba Ortega-Gascó 1,2,† , Marc Hernaiz-Llorens 1,2 , Maria Antonia Odena 4, Fausto Ulloa 1,2, Eliandre de Oliveira 4, Miquel Bosch 3 , Lluís Pujadas 1,2 and Eduardo Soriano 1,2,* 1 Department of Cell Biology, Physiology and Immunology, University of Barcelona and Institute of Neurosciences, 08028 Barcelona, Spain; [email protected] (A.O.-G.); [email protected] (M.H.-L.); [email protected] (F.U.); [email protected] (L.P.) 2 Centro de Investigación Biomédica en Red sobre Enfermedades Neurodegenerativas (CIBERNED), 28031 Madrid, Spain 3 Department of Basic Sciences, Universitat Internacional de Catalunya, 08195 Sant Cugat del Vallès, Spain; [email protected] 4 Plataforma de Proteòmica, Parc Científic de Barcelona (PCB), 08028 Barcelona, Spain; [email protected] (M.A.O.); [email protected] (E.d.O.) * Correspondence: [email protected] (A.P.); [email protected] (E.S.) † A.P. and A.O.-G. contributed equally. Abstract: Neuronal cell adhesion molecule 2 (NCAM2) is a membrane protein with an important role in the morphological development of neurons. In the cortex and the hippocampus, NCAM2 is essential for proper neuronal differentiation, dendritic and axonal outgrowth and synapse forma- tion. However, little is known about NCAM2 functional mechanisms and its interactive partners Citation: Parcerisas, A.; during brain development. Here we used mass spectrometry to study the molecular interactome Ortega-Gascó, A.; Hernaiz-Llorens, of NCAM2 in the second postnatal week of the mouse cerebral cortex. -

Deciphering the Molecular Profile of Plaques, Memory Decline And
ORIGINAL RESEARCH ARTICLE published: 16 April 2014 AGING NEUROSCIENCE doi: 10.3389/fnagi.2014.00075 Deciphering the molecular profile of plaques, memory decline and neuron loss in two mouse models for Alzheimer’s disease by deep sequencing Yvonne Bouter 1†,Tim Kacprowski 2,3†, Robert Weissmann4, Katharina Dietrich1, Henning Borgers 1, Andreas Brauß1, Christian Sperling 4, Oliver Wirths 1, Mario Albrecht 2,5, Lars R. Jensen4, Andreas W. Kuss 4* andThomas A. Bayer 1* 1 Division of Molecular Psychiatry, Georg-August-University Goettingen, University Medicine Goettingen, Goettingen, Germany 2 Department of Bioinformatics, Institute of Biometrics and Medical Informatics, University Medicine Greifswald, Greifswald, Germany 3 Department of Functional Genomics, Interfaculty Institute for Genetics and Functional Genomics, University Medicine Greifswald, Greifswald, Germany 4 Human Molecular Genetics, Department for Human Genetics of the Institute for Genetics and Functional Genomics, Institute for Human Genetics, University Medicine Greifswald, Ernst-Moritz-Arndt University Greifswald, Greifswald, Germany 5 Institute for Knowledge Discovery, Graz University of Technology, Graz, Austria Edited by: One of the central research questions on the etiology of Alzheimer’s disease (AD) is the Isidro Ferrer, University of Barcelona, elucidation of the molecular signatures triggered by the amyloid cascade of pathological Spain events. Next-generation sequencing allows the identification of genes involved in disease Reviewed by: Isidro Ferrer, University of Barcelona, processes in an unbiased manner. We have combined this technique with the analysis of Spain two AD mouse models: (1) The 5XFAD model develops early plaque formation, intraneu- Dietmar R. Thal, University of Ulm, ronal Ab aggregation, neuron loss, and behavioral deficits. (2)TheTg4–42 model expresses Germany N-truncated Ab4–42 and develops neuron loss and behavioral deficits albeit without plaque *Correspondence: formation. -

Novel Mechanistic Targets of Forkhead Box Q1 Transcription Factor in Human Breast Cancer Cells
bioRxiv preprint doi: https://doi.org/10.1101/2020.05.26.117176; this version posted May 29, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Novel mechanistic targets of Forkhead box Q1 transcription factor in human breast cancer cells Su-Hyeong Kim*,1, Eun-Ryeong Hahm*,1, Krishna B. Singh*, and Shivendra V. Singh*,¶,2 From the *Department of Pharmacology & Chemical Biology, and ¶UPMC Hillman Cancer Center, University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania Running Title: Role of FoxQ1 in breast cancer 2To whom correspondence should be addressed: 2.32A Hillman Cancer Center Research Pavilion, UPMC Hillman Cancer Center, 5117 Centre Avenue, Pittsburgh, PA 15213. Phone: 412-623-3263; Fax: 412-623-7828; E-mail: [email protected] Keywords: Breast Cancer, FoxQ1, Cell Cycle, Interleukin-1α, Interleukin-8 The transcription factor forkhead box Q1 analysis. FoxQ1 overexpression resulted (FoxQ1), which is overexpressed in in downregulation of genes associated different solid tumors, has emerged as a with cell cycle checkpoints, M phase, and key player in the pathogenesis of breast cellular response to stress/external stimuli cancer by regulating epithelial- as evidenced from the Reactome pathway mesenchymal transition, maintenance of analysis. Consequently, FoxQ1 cancer-stem like cells, and metastasis. overexpression resulted in S, G2M and However, the mechanism underlying mitotic arrest in basal-like SUM159 and oncogenic function of FoxQ1 is still not HMLE cells, but not in luminal-type fully understood. In this study, we MCF-7 cells. There were differences in compared the RNA-seq data from FoxQ1 expression of cell cycle-associated overexpressing SUM159 cells with that of proteins between FoxQ1 overexpressing empty vector-transfected control (EV) SUM159 and MCF-7 cells. -

Host Cell Factors Necessary for Influenza a Infection: Meta-Analysis of Genome Wide Studies
Host Cell Factors Necessary for Influenza A Infection: Meta-Analysis of Genome Wide Studies Juliana S. Capitanio and Richard W. Wozniak Department of Cell Biology, Faculty of Medicine and Dentistry, University of Alberta Abstract: The Influenza A virus belongs to the Orthomyxoviridae family. Influenza virus infection occurs yearly in all countries of the world. It usually kills between 250,000 and 500,000 people and causes severe illness in millions more. Over the last century alone we have seen 3 global influenza pandemics. The great human and financial cost of this disease has made it the second most studied virus today, behind HIV. Recently, several genome-wide RNA interference studies have focused on identifying host molecules that participate in Influen- za infection. We used nine of these studies for this meta-analysis. Even though the overlap among genes identified in multiple screens was small, network analysis indicates that similar protein complexes and biological functions of the host were present. As a result, several host gene complexes important for the Influenza virus life cycle were identified. The biological function and the relevance of each identified protein complex in the Influenza virus life cycle is further detailed in this paper. Background and PA bound to the viral genome via nucleoprotein (NP). The viral core is enveloped by a lipid membrane derived from Influenza virus the host cell. The viral protein M1 underlies the membrane and anchors NEP/NS2. Hemagglutinin (HA), neuraminidase Viruses are the simplest life form on earth. They parasite host (NA), and M2 proteins are inserted into the envelope, facing organisms and subvert the host cellular machinery for differ- the viral exterior. -

Myosin VB in Intestinal Pathogenesis Fernando Cartón García
ADVERTIMENT. Lʼaccés als continguts dʼaquesta tesi queda condicionat a lʼacceptació de les condicions dʼús establertes per la següent llicència Creative Commons: http://cat.creativecommons.org/?page_id=184 ADVERTENCIA. El acceso a los contenidos de esta tesis queda condicionado a la aceptación de las condiciones de uso establecidas por la siguiente licencia Creative Commons: http://es.creativecommons.org/blog/licencias/ WARNING. The access to the contents of this doctoral thesis it is limited to the acceptance of the use conditions set by the following Creative Commons license: https://creativecommons.org/licenses/?lang=en Myosin VB in intestinal pathogenesis Doctoral thesis presented by Fernando Cartón García For the PhD degree Biochemistry, Molecular Biology and Biomedicine doctorate program Biochemistry and Molecular Biology Department Faculty of Medicine Universidad Autónoma de Barcelona Work carried out in Group of Biomedical Research in Digestive Tract Tumors Molecular Biology and Biochemistry Research Center (CIBBIM-Nanomedicine) Vall d’Hebron Research Institute June 2017 DIRECTOR ACADEMIC TUTOR Dr. Diego Arango Dr. Joan Seoane PhD STUDENT Fernando Cartón 2 INDEX RESUMEN ................................................................................. 6 ABSTRACT ............................................................................... 8 INTRODUCTION ..................................................................... 10 1. The human digestive tract .................................................. 10 1.1 Anatomy and Histology -

Proteomic Signatures of Brain Regions Affected by Tau Pathology in Early and Late Stages of Alzheimer's Disease
Neurobiology of Disease 130 (2019) 104509 Contents lists available at ScienceDirect Neurobiology of Disease journal homepage: www.elsevier.com/locate/ynbdi Proteomic signatures of brain regions affected by tau pathology in early and T late stages of Alzheimer's disease Clarissa Ferolla Mendonçaa,b, Magdalena Kurasc, Fábio César Sousa Nogueiraa,d, Indira Plác, Tibor Hortobágyie,f,g, László Csibae,h, Miklós Palkovitsi, Éva Renneri, Péter Dömej,k, ⁎ ⁎ György Marko-Vargac, Gilberto B. Domonta, , Melinda Rezelic, a Proteomics Unit, Department of Biochemistry, Federal University of Rio de Janeiro, Rio de Janeiro, Brazil b Gladstone Institute of Neurological Disease, San Francisco, USA c Division of Clinical Protein Science & Imaging, Department of Clinical Sciences (Lund) and Department of Biomedical Engineering, Lund University, Lund, Sweden d Laboratory of Proteomics, LADETEC, Institute of Chemistry, Federal University of Rio de Janeiro, Rio de Janeiro, Brazil e MTA-DE Cerebrovascular and Neurodegenerative Research Group, University of Debrecen, Debrecen, Hungary f Institute of Pathology, Faculty of Medicine, University of Szeged, Szeged, Hungary g Centre for Age-Related Medicine, SESAM, Stavanger University Hospital, Stavanger, Norway h Department of Neurology, Faculty of Medicine, University of Debrecen, Debrecen, Hungary i SE-NAP – Human Brain Tissue Bank Microdissection Laboratory, Semmelweis University, Budapest, Hungary j Department of Psychiatry and Psychotherapy, Semmelweis University, Budapest, Hungary k National Institute of Psychiatry and Addictions, Nyírő Gyula Hospital, Budapest, Hungary ARTICLE INFO ABSTRACT Keywords: Background: Alzheimer's disease (AD) is the most common neurodegenerative disorder. Depositions of amyloid β Alzheimer's disease peptide (Aβ) and tau protein are among the major pathological hallmarks of AD. Aβ and tau burden follows Proteomics predictable spatial patterns during the progression of AD.