Hybrid Inhibitors of DNA Gyrase a and B: Design, Synthesis and Evaluation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Intravenous Palonosetron Increases the Incidence of Qtc Prolongation During Sevoflurane General Anesthesia for Laparotomy
Korean J Anesthesiol 2013 November 65(5): 397-402 Clinical Research Article http://dx.doi.org/10.4097/kjae.2013.65.5.397 Intravenous palonosetron increases the incidence of QTc prolongation during sevoflurane general anesthesia for laparotomy Jeong Jin Min, Yongjae Yoo, Tae Kyong Kim, and Jung-Man Lee Department of Anesthesiology and Pain Medicine, Seoul National University Hospital, Seoul National University College of Medicine, Seoul, Korea Background: Palonosetron is a recently introduced 5-hydroxytryptamine-3 (5-HT3) receptor antagonist useful for postoperative nausea and vomiting prophylaxis. However, 5-HT3 receptor antagonists increase the corrected QT (QTc) interval in patients who undergo general anesthesia. This retrospective study was performed to evaluate whether palono- setron would induce a QTc prolongation in patients undergoing general anesthesia with sevoflurane. Methods: We reviewed a database of 81 patients who underwent general anesthesia with sevoflurane. We divided the records into palonosetron (n = 41) and control (n = 40) groups according to the use of intraoperative palonosetron, and analyzed the electrocardiographic data before anesthesia and 30, 60, 90, and 120 min after skin incision. Changes in the QTc interval from baseline, mean blood pressure, heart rate, body temperature, and sevoflurane concentrations at each time point were compared between the two groups. Results: The QTc intervals at skin incision, and 30, 60, 90, and 120 min after the skin incision during general anesthesia were significantly longer than those at baseline in the two groups (P < 0.001). The changes in the QTc intervals were not different between the two groups (P = 0.41). However, six patients in the palonosetron group showed a QTc interval > 500 ms 30 min after skin incision, whereas no patient did in the control group (P = 0.01). -

Antibacterial Effect of Sevoflurane and Isoflurane Ángel Martínez-Monsalve3 María Dolores Crespo- Sánchez1
Original María Martínez-Serrano1 Manuel Gerónimo-Pardo2 Antibacterial effect of sevoflurane and isoflurane Ángel Martínez-Monsalve3 María Dolores Crespo- Sánchez1 1Servicio de Microbiología y Parasitología. Complejo Hospitalario Universitario de Albacete. 2Servicio de Anestesiología y Reanimación. Complejo Hospitalario Universitario de Albacete. 3Servicio de Cirugía Vascular. Complejo Hospitalario Universitario de Albacete. ABSTRACT property in vivo. This might then allow these agents to be con- sidered as rescue treatment against multidrug resistant patho- Introduction. Multidrug resistant bacteria are increasing gens, including a topical use in infected wounds. worldwide and therapeutic options are limited. Some anaes- Key words: Sevoflurane, Isoflurane, Anaesthetics, Inhala- thetics have shown antibacterial activity before. In this study, tion, Anti-Infective Agents. we have investigated the antibacterial effect of the halogen- ated anaesthetic agents sevoflurane and isoflurane against a range of resistant pathogens. Actividad antibacteriana de sevoflurano e Methods. Two experiments were conducted. In the first, isoflurano bacterial suspensions of both ATCC and resistant strains of Staphylococcus aureus, Escherichia coli and Pseudomonas RESUMEN aeruginosa were exposed to liquid sevoflurane and isoflurane during 15, 30 and 60 minutes. In the second experiment clinical Introducción. Las bacterias multirresistentes están au- resistant strains of E. coli, Klebsiella pneumoniae, Enterobac- mentando en todo el mundo y las opciones terapéuticas son ter cloacae, P. aeruginosa, Acinetobacter baumannii, S. aureus, limitadas. Algunos anestésicos han mostrado actividad an- and Enterococcus faecium were studied. Previously inoculated tibacteriana previamente. En este estudio hemos investigado agar plates were irrigated with the halogenated anaesthet- dicha actividad en los anestésicos halogenados sevoflurano e ic agents and these were left to evaporate before the plates isoflurano frente a un grupo de patógenos resistentes. -

Antibacterial Activity and Mechanisms of Essential Oil from Citrus Medica L
molecules Article Antibacterial Activity and Mechanisms of Essential Oil from Citrus medica L. var. sarcodactylis Ze-Hua Li 1,2, Ming Cai 2,*, Yuan-Shuai Liu 3, Pei-Long Sun 2,* and Shao-Lei Luo 2 1 College of Biotechnology and Bioengineering, Zhejiang University of Technology, Hangzhou 310014, China; [email protected] 2 Department of Food Science and Technology, Zhejiang University of Technology, Hangzhou 310014, China; [email protected] 3 Department of Chemical and Biological Engineering, Hong Kong University of Science and Technology, Hong Kong; [email protected] * Correspondence: [email protected] (P.-L.S.); [email protected] (M.C.); Tel.: +86-0571-88320388 (P.-L.S.); +86-0571-88320345 (M.C.) Academic Editor: Francesca Mancianti Received: 29 March 2019; Accepted: 18 April 2019; Published: 22 April 2019 Abstract: In this work, antibacterial activity of finger citron essential oil (FCEO, Citrus medica L. var. sarcodactylis) and its mechanism against food-borne bacteria were evaluated. A total of 28 components in the oil were identified by gas chromatography-mass spectrometry, in which limonene (45.36%), γ-terpinene (21.23%), and dodecanoic acid (7.52%) were three main components. For in vitro antibacterial tests, FCEO exhibited moderately antibacterial activity against common food-borne bacteria: Escherichia coli, Staphylococcus aureus, Bacillus subtilis and Micrococcus luteus. It showed a better bactericidal effect on Gram-positive bacteria than Gram-negative. Mechanisms of the antibacterial action were investigated by observing changes of bacteria morphology according to scanning electron microscopy, time-kill analysis, and permeability of cell and membrane integrity. Morphology of tested bacteria was changed and damaged more seriously with increased concentration and exposure time of FCEO. -

(12) Patent Application Publication (10) Pub. No.: US 2014/0296.191 A1 PATEL Et Al
US 20140296.191A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2014/0296.191 A1 PATEL et al. (43) Pub. Date: Oct. 2, 2014 (54) COMPOSITIONS OF PHARMACEUTICAL (52) U.S. Cl. ACTIVES CONTAINING DETHYLENE CPC ............... A61K 47/10 (2013.01); A61 K9/0019 GLYCOL MONOETHYLETHER OR OTHER (2013.01); A61 K9/0048 (2013.01); A61 K ALKYL DERVATIVES 45/06 (2013.01) USPC ........... 514/167: 514/177; 514/178: 514/450; (71) Applicant: THEMIS MEDICARE LIMITED, 514/334: 514/226.5: 514/449; 514/338; Mumbai (IN) 514/256; 514/570; 514/179; 514/174: 514/533; (72) Inventors: Dinesh Shantilal PATEL, Mumbai (IN); 514/629; 514/619 Sachin Dinesh PATEL, Mumbai (IN); Shashikant Prabhudas KURANI, Mumbai (IN); Madhavlal Govindlal (57) ABSTRACT PATEL, Mumbai (IN) (73) Assignee: THEMIS MEDICARE LIMITED, The present invention relates to pharmaceutical compositions Mumbai (IN) of various pharmaceutical actives, especially lyophilic and hydrophilic actives containing Diethylene glycol monoethyl (21) Appl. No.: 14/242,973 ether or other alkyl derivatives thereofas a primary vehicle and/or to pharmaceutical compositions utilizing Diethylene (22) Filed: Apr. 2, 2014 glycol monoethyl ether or other alkyl derivatives thereofas a primary vehicle or as a solvent system in preparation of Such (30) Foreign Application Priority Data pharmaceutical compositions. The pharmaceutical composi Apr. 2, 2013 (IN) ......................... 1287/MUMA2013 tions of the present invention are safe, non-toxic, exhibits enhanced physical stability compared to conventional formu Publication Classification lations containing such pharmaceutical actives and are Suit able for use as injectables for intravenous and intramuscular (51) Int. Cl. administration, as well as for use as a preformed solution/ A647/ (2006.01) liquid for filling in and preparation of capsules, tablets, nasal A6 IK 45/06 (2006.01) sprays, gargles, dermal applications, gels, topicals, liquid oral A6 IK9/00 (2006.01) dosage forms and other dosage forms. -

Jp Xvii the Japanese Pharmacopoeia
JP XVII THE JAPANESE PHARMACOPOEIA SEVENTEENTH EDITION Official from April 1, 2016 English Version THE MINISTRY OF HEALTH, LABOUR AND WELFARE Notice: This English Version of the Japanese Pharmacopoeia is published for the convenience of users unfamiliar with the Japanese language. When and if any discrepancy arises between the Japanese original and its English translation, the former is authentic. The Ministry of Health, Labour and Welfare Ministerial Notification No. 64 Pursuant to Paragraph 1, Article 41 of the Law on Securing Quality, Efficacy and Safety of Products including Pharmaceuticals and Medical Devices (Law No. 145, 1960), the Japanese Pharmacopoeia (Ministerial Notification No. 65, 2011), which has been established as follows*, shall be applied on April 1, 2016. However, in the case of drugs which are listed in the Pharmacopoeia (hereinafter referred to as ``previ- ous Pharmacopoeia'') [limited to those listed in the Japanese Pharmacopoeia whose standards are changed in accordance with this notification (hereinafter referred to as ``new Pharmacopoeia'')] and have been approved as of April 1, 2016 as prescribed under Paragraph 1, Article 14 of the same law [including drugs the Minister of Health, Labour and Welfare specifies (the Ministry of Health and Welfare Ministerial Notification No. 104, 1994) as of March 31, 2016 as those exempted from marketing approval pursuant to Paragraph 1, Article 14 of the Same Law (hereinafter referred to as ``drugs exempted from approval'')], the Name and Standards established in the previous Pharmacopoeia (limited to part of the Name and Standards for the drugs concerned) may be accepted to conform to the Name and Standards established in the new Pharmacopoeia before and on September 30, 2017. -

2016 Medicines in Development for Rare Diseases a LIST of ORPHAN DRUGS in the PIPELINE
2016 Medicines in Development for Rare Diseases A LIST OF ORPHAN DRUGS IN THE PIPELINE Autoimmune Diseases Product Name Sponsor Official FDA Designation* Development Status Actemra® Genentech treatment of systemic sclerosis Phase III tocilizumab South San Francisco, CA www.gene.com Adempas® Bayer HealthCare Pharmaceuticals treatment of systemic sclerosis Phase II riociguat Whippany, NJ www.pharma.bayer.com ARA 290 Araim Pharmaceuticals treatment of neuropathic pain in patients Phase II Tarrytown, NY with sarcoidosis www.ariampharma.com ARG201 arGentis Pharmaceuticals treatment of diffuse systemic sclerosis Phase II (type 1 native bovine skin Collierville, TN www.argentisrx.com collagen) BYM338 Novartis Pharmaceuticals treatment of inclusion body myositis Phase III (bimagrumab) East Hanover, NJ www.novartis.com CCX168 ChemoCentryx treatment of anti-neutrophil cytoplasmic Phase II (5a receptor antagonist) Mountain View, CA auto-antibodies associated vasculitides www.chemocentryx.com (granulomatosis with polyangitis or Wegener's granulomatosis), microscopic polyangitis, and Churg-Strauss syndrome * This designation is issued by the FDA's Office of Orphan Products Development while the drug is still in development. The designation makes the sponsor of the drug eligible for entitlements under the Orphan Drug Act of 1983. The entitlements include seven years of marketing exclusivity following FDA approval of the drug for the designated use. Medicines in Development: Rare Diseases | 2016 1 Autoimmune Diseases Product Name Sponsor Official FDA -
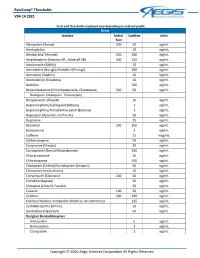
Paincomp® Thresholds V04.14.2021 Copyright © 2020 Aegis Sciences
PainComp® Thresholds V04.14.2021 Tests and thresholds employed vary depending on ordered profile Urine Analyte Initial Confirm Units Test Alprazolam (Xanax) 200 50 ng/mL Amitriptyline 10 ng/mL Amobarbital (Amytal) 200 200 ng/mL Amphetamine (Adzenys ER , Adderall XR) 500 250 ng/mL Aripiprazole (Abilify) 10 ng/mL Armodafinil (Nuvigil)/Modafinil (Provigil) 100 ng/mL Asenapine (Saphris) 10 ng/mL Atomoxetine (Strattera) 10 ng/mL Baclofen 500 ng/mL Benzodiazepines (Chlordiazepoxide, Clorazepate, 200 50 ng/mL Diazepam, Oxazepam, Temazepam) Brexpiprazole (Rexulti) 10 ng/mL Buprenorphine/Sublingual (Belbuca) 1 ng/mL Buprenorphine/Transdermal patch (Butrans) 1 ng/mL Bupropion (Aplenzin, Forfivo XL) 50 ng/mL Buspirone 25 ng/mL Butalbital 200 200 ng/mL Butorphanol 1 ng/mL Caffeine 15 mcg/mL Carbamazepine 50 ng/mL Cariprazine (Vraylar) 20 ng/mL Carisoprodol (Soma)/Meprobamate 200 ng/mL Chlorpromazine 10 ng/mL Chlorzoxazone 500 ng/mL Citalopram (Celexa)/Escitalopram (Lexapro) 50 ng/mL Clomipramine (Anafranil) 10 ng/mL Clonazepam (Klonopin) 200 50 ng/mL Clonidine (Kapvay) 50 ng/mL Clozapine (Clozaril, Fazaclo) 50 ng/mL Cocaine 100 50 ng/mL Codeine 200 100 ng/mL Cotinine/Nicotine metabolite (Habitrol, Nicoderm CQ) 125 ng/mL Cyclobenzaprine (Amrix) 10 ng/mL Dantrolene (Dantrium) 50 ng/mL Designer Benzodiazepines Adinazolam 5 ng/mL Bromazolam 1 ng/mL Clonazolam 1 ng/mL Copyright © 2020 Aegis Sciences Corporation All Rights Reserved PainComp® Thresholds V04.14.2021 Deschloroetizolam 1 ng/mL Diclazepam 1 ng/mL Etizolam 1 ng/mL Flualprazolam 1 ng/mL Flubromazepam -

Bulk Drug Substances Nominated for Use in Compounding Under Section 503B of the Federal Food, Drug, and Cosmetic Act
Updated June 07, 2021 Bulk Drug Substances Nominated for Use in Compounding Under Section 503B of the Federal Food, Drug, and Cosmetic Act Three categories of bulk drug substances: • Category 1: Bulk Drug Substances Under Evaluation • Category 2: Bulk Drug Substances that Raise Significant Safety Risks • Category 3: Bulk Drug Substances Nominated Without Adequate Support Updates to Categories of Substances Nominated for the 503B Bulk Drug Substances List1 • Add the following entry to category 2 due to serious safety concerns of mutagenicity, cytotoxicity, and possible carcinogenicity when quinacrine hydrochloride is used for intrauterine administration for non- surgical female sterilization: 2,3 o Quinacrine Hydrochloride for intrauterine administration • Revision to category 1 for clarity: o Modify the entry for “Quinacrine Hydrochloride” to “Quinacrine Hydrochloride (except for intrauterine administration).” • Revision to category 1 to correct a substance name error: o Correct the error in the substance name “DHEA (dehydroepiandosterone)” to “DHEA (dehydroepiandrosterone).” 1 For the purposes of the substance names in the categories, hydrated forms of the substance are included in the scope of the substance name. 2 Quinacrine HCl was previously reviewed in 2016 as part of FDA’s consideration of this bulk drug substance for inclusion on the 503A Bulks List. As part of this review, the Division of Bone, Reproductive and Urologic Products (DBRUP), now the Division of Urology, Obstetrics and Gynecology (DUOG), evaluated the nomination of quinacrine for intrauterine administration for non-surgical female sterilization and recommended that quinacrine should not be included on the 503A Bulks List for this use. This recommendation was based on the lack of information on efficacy comparable to other available methods of female sterilization and serious safety concerns of mutagenicity, cytotoxicity and possible carcinogenicity in use of quinacrine for this indication and route of administration. -

WHO Drug Information Vol
WHO Drug Information Vol. 34, No. 2, 2020 WHO Drug Information Contents Publication News 143 Publication of the 54th report of the World Health Organization (WHO) Expert Committee on Specifications for Pharmaceutical Preparations (ECSPP) (WHO Technical Series, N° 1025) Consultation Documents 146 Points to consider on the different approaches – including HBEL – to establish carryover limits in cleaning validation for identification of contamination risks when manufacturing in shared facilities 164 World Health Organization/United Nations Population Fund recommendations for condom storage and shipping 170 World Health Organization/United Nations Population Fund Guidance on conducting post-market surveillance of condoms 179 World Health Organization/United Nations Population Fund Guidance on testing of male latex condoms 201 Good reliance practices in regulatory decision-making: high-level principles and recommendations 231 Guideline on data integrity ATC/DDD Classification 255 ATC/DDD Classification (Temporary) 261 ATC/DDD Classification (Final) International Nonproprietary Names (INN) 263 List No. 123 of Proposed International Nonproprietary Names (INN) for Pharmaceutical Substances Abbreviations and websites CHMP Committee for Medicinal Products for Human Use (EMA) EMA European Medicines Agency (www.ema.europa.eu) EU European Union FDA U.S. Food and Drug Administration (www.fda.gov) Health Canada Federal department responsible for health product regulation in Canada (www.hc-sc.gc.ca) HPRA Health Products Regulatory Authority, Ireland -

2003 AAZV Proceedings.Pdf
COMPARISON OF ISOFLURANE AND SEVOFLURANE ANESTHESIA FOLLOWING PREMEDICATION WITH BUTORPHANOL IN THE GREEN IGUANA (Iguana iguana) Sonia M. Hernandez-Divers, D VM, Dipl ACZM,' * Juergen Schumacher, Dr.med. vet., Dipl ACZM,' Matt R. Read, DVM, MVSc, Dip1 ACVA,' Scott Stahl, D VM, AB VP (A~ian),~and Stephen Hernandez-Divers, B VetMed, DZooMed, MRCVS' 'Exotic Animal, Wildlge and Zoological Medicine, Department of Small Animal Medicine and Surgery, College of Veterinary Medicine, University of Georgia, Athens, GA 30602 USA; 'Department of Small Animal Clinical Sciences, College of Veterinary Medicine, The University of Tennessee, Knoxville, TN 3 7996 USA; 'Anesthesiology, Department of Small Animal Medicine and Surgery, College of Veterinary Medicine, University of Georgia, Athens, GA 30602 USA; 'Eastern Exotics Center, 4001 Legato Road, Fairfa, VA 22033 USA Abstract Twenty-three, male captive-bred green iguanas (Iguana iguana) weighing 0.5 1-0.67 kg were anesthetized to determine the effectiveness and the cardiopulmonary effects of butorphanol- sevoflurane and butorphanol-isoflurane. Baseline heart and respiratory rates were recorded on all animals prior to administration of intramuscular butorphanol (2 mgikg). Thirty minutes after premedication, the heart rate and respiratory rate of each iguana was recorded and 12 animals were induced with isoflurane (5% in 100% oxygen), and 11 were induced with sevoflurane (8% in 100% oxygen), via face mask. Following the loss of the righting reflex, each iguana was intubated and intermittent positive pressure ventilation (IPPV) was provided at 10-12 breathdmin with a small animal ventilator. Mean maintenance isoflurane and sevoflurane concentrations were 2% and 4%, respectively. Heart rate, functional oxygen hemoglobin saturation (SpO,), and end-tidal CO, concentrations were measured every 5 min. -

A Comparison of the Effect of Sevoflurane and Propofol on Ventricular Repolarisation After Preoperative Cefuroxime Infusion
Hindawi BioMed Research International Volume 2019, Article ID 8978906, 6 pages https://doi.org/10.1155/2019/8978906 Research Article A Comparison of the Effect of Sevoflurane and Propofol on Ventricular Repolarisation after Preoperative Cefuroxime Infusion Yanqiu Liu,1 Hong Gao ,1 Guilong Wang ,2 Li An,1 Jing Yi,2 Xiaokui Fu,2 Chunlei Wen,2 and Zijun Wang2 1 Department of Anesthesiology, Te Afliated Hospital of Guizhou Medical University, Guiyang, Guizhou, China 2Department of Anesthesiology, Guizhou Medical University, Guiyang, Guizhou, China Correspondence should be addressed to Hong Gao; [email protected] Received 10 July 2018; Revised 11 December 2018; Accepted 13 December 2018; Published 2 January 2019 Academic Editor: Gianluca Di Bella Copyright © 2019 Yanqiu Liu et al. Tis is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Te aim of this study is to investigate the changes in QT, QTc, and Tp-e intervals and Tp-e/QT ratio on surface electrocardiogram (ECG) signals during anaesthesia induction using propofol or sevofurane afer preoperative cefuroxime infusion. Some 120 cases of gynaecological patients are randomly divided into propofol (P) and sevofurane (S) groups (n=60). Afer cefuroxime (1.5 g) was infused in the two groups of patients, propofol target controlled infusion (TCI) was conducted in the P group for 5 min to realise a plasma concentration of 4 �g/ml while patients in the S group inhaled anaesthesia by infusing 1.3 MAC sevofurane for 6 min. Te 12-lead ECGs were separately collected before infusing cefuroxime (T1), afer infusing cefuroxime (T2), and afer infusing propofol or sevofurane (T3) to measure QT and Tp-e intervals, calculate QTc and Tp-e/QT, and record MAP and HR. -

Pocket Anesthesia Reference Card
OBSTETRICS & OB EMERGENCIES OBSTETRICS & OB EMERGENCIES NEURAXIAL ANESTHESIA EMERGENCIES Pocket Anesthesia (Please see full OB pocket card for details) (Please see full OB pocket card for details) v 0.9 Key Points High Spinal & Total Spinal Hypertensive Disorders Urgent or Emergent C-Section & Emergent GA Reference Card •Uses: C/S, Gyn, Uro, Abdo & LE procedures Signs •Numbness, paresthesia, or weakness of UE’s Pre-Eclampsia: BP > 140/90 x2 ≥ 20 wks, proteinuria, +/- organ dysfunct. For all: Pre-induction checklist •High spinal is a significant cause of morbidity/mortality → see emergencies •Rapid unexpected rise of sensory block Card design by providers from many institutions including: •Consider delivery •Call for help, take AMPLE Hx, IV access, NaCit, pulse ox, LUD. •Monitor BP q1-5 min before, during, & after. Use standard monitors •SOB, apnea, bradycardia, hypotension, or nausea/vomiting •Prevent seizure: Mg 4-6 g IV over 15-20 min + 1-2 g/hr gtt for 24 hr post •Neuraxial preferred if time - plan determined by degree of urgency, •Ensure adequate IV access, vasoconstrictors & GA available •Loss of consciousness (LOC = total spinal), Cardiac arrest delivery (do NOT d/c in OR); (10 g IM load described if no PIV) communication w/OB team, resources, & pt. condition •Consider preloading with IVF (Avoid in pre-eclampsia) • •Tx severe HTN (SBP > 155, DBP > 105): 1st line: Labetalol IV, hydralazine IV, • If CS for fetal distress, ↑ O2 to baby: SPOILT-Stop oxytocin, Position-LUD, O2, IV fluid, Low Tx Call for help & code cart, inform team •Consider starting vasopressor support at time of placement •If cardiac arrest: start CPR, refer to ACLS protocol nifedipine PO and no IV (others okay if 1st line unavailable) BP (give pressor), Tocolytics (terbutaline 250 mcg subQ, +/-NTG SL spray 400 mcg x2) •Ensure aseptic technique for placement •Watch for Mg tox: ↓ DTRs, Resp/cardiac comp.