Research Article DNA AS EVIDENCE for the EXISTENCE of RELICT
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Urban Myths Mythical Cryptids
Ziptales Advanced Library Worksheet 2 Urban Myths Mythical Cryptids ‘What is a myth? It is a story that pretends to be real, but is in fact unbelievable. Like many urban myths it has been passed around (usually by word of mouth), acquiring variations and embellishments as it goes. It is a close cousin of the tall tale. There are mythical stories about almost any aspect of life’. What do we get when urban myths meet the animal kingdom? We find a branch of pseudoscience called cryptozoology. Cryptozoology refers to the study of and search for creatures whose existence has not been proven. These creatures (or crytpids as they are known) appear in myths and legends or alleged sightings. Some examples include: sea serpents, phantom cats, unicorns, bunyips, giant anacondas, yowies and thunderbirds. Some have even been given actual names you may have heard of – do Yeti, Owlman, Mothman, Cyclops, Bigfoot and the Loch Ness Monster sound familiar? Task 1: Choose one of the cryptids from the list above (or perhaps one that you may already know of) and write an informative text identifying the following aspects of this mythical creature: ◊ Description ◊ Features ◊ Location ◊ First Sighting ◊ Subsequent Sightings ◊ Interesting Facts (e.g. how is it used in popular culture? Has it been featured in written or visual texts?) Task 2: Cryptozoologists claim there have been cases where species now accepted by the scientific community were initially considered urban myths. Can you locate any examples of creatures whose existence has now been proven but formerly thought to be cryptids? Extension Activities: • Cryptozoology is called a ‘pseudoscience’ because it relies solely on anecdotes and reported sightings rather than actual evidence. -

Book Section Reprint the STRUGGLE for TROGLODYTES1
The RELICT HOMINOID INQUIRY 6:33-170 (2017) Book Section Reprint THE STRUGGLE FOR TROGLODYTES1 Boris Porshnev "I have no doubt that some fact may appear fantastic and incredible to many of my readers. For example, did anyone believe in the existence of Ethiopians before seeing any? Isn't anything seen for the first time astounding? How many things are thought possible only after they have been achieved?" (Pliny, Natural History of Animals, Vol. VII, 1) INTRODUCTION BERNARD HEUVELMANS Doctor in Zoological Sciences How did I come to study animals, and from the study of animals known to science, how did I go on to that of still undiscovered animals, and finally, more specifically to that of unknown humans? It's a long story. For me, everything started a long time ago, so long ago that I couldn't say exactly when. Of course it happened gradually. Actually – I have said this often – one is born a zoologist, one does not become one. However, for the discipline to which I finally ended up fully devoting myself, it's different: one becomes a cryptozoologist. Let's specify right now that while Cryptozoology is, etymologically, "the science of hidden animals", it is in practice the study and research of animal species whose existence, for lack of a specimen or of sufficient anatomical fragments, has not been officially recognized. I should clarify what I mean when I say "one is born a zoologist. Such a congenital vocation would imply some genetic process, such as that which leads to a lineage of musicians or mathematicians. -

The Sherpa and the Snowman
THE SHERPA AND THE SNOWMAN Charles Stonor the "Snowman" exist an ape DOESlike creature dwelling in the unexplored fastnesses of the Himalayas or is he only a myth ? Here the author describes a quest which began in the foothills of Nepal and led to the lower slopes of Everest. After five months of wandering in the vast alpine stretches on the roof of the world he and his companions had to return without any demon strative proof, but with enough indirect evidence to convince them that the jeti is no myth and that one day he will be found to be of a a very remarkable man-like ape type thought to have died out thousands of years before the dawn of history. " Apart from the search for the snowman," the narrative investigates every aspect of life in this the highest habitable region of the earth's surface, the flora and fauna of the little-known alpine zone below the snow line, the unexpected birds and beasts to be met with in the Great Himalayan Range, the little Buddhist communities perched high up among the crags, and above all the Sherpas themselves that stalwart people chiefly known to us so far for their gallant assistance in climbing expeditions their yak-herding, their happy family life, and the wav they cope with the bleak austerity of their lot. The book is lavishly illustrated with the author's own photographs. THE SHERPA AND THE SNOWMAN "When the first signs of spring appear the Sherpas move out to their grazing grounds, camping for the night among the rocks THE SHERPA AND THE SNOWMAN By CHARLES STONOR With a Foreword by BRIGADIER SIR JOHN HUNT, C.B.E., D.S.O. -

The Aquatic Ape Hypothesis: Most Credible Theory of Human Evolution Free Download
THE AQUATIC APE HYPOTHESIS: MOST CREDIBLE THEORY OF HUMAN EVOLUTION FREE DOWNLOAD Elaine Morgan | 208 pages | 01 Oct 2009 | Souvenir Press Ltd | 9780285635180 | English | London, United Kingdom Aquatic ape hypothesis In addition, the evidence cited by AAH The Aquatic Ape Hypothesis: Most Credible Theory of Human Evolution mostly concerned developments in soft tissue anatomy and physiology, whilst paleoanthropologists rarely speculated on evolutionary development of anatomy beyond the musculoskeletal system and brain size as revealed in fossils. His summary at the end was:. From Wikipedia, the free encyclopedia. Proceedings of the National Academy of Sciences. Thanks for your comment! List of individual apes non-human Apes in space non-human Almas Bigfoot Bushmeat Chimpanzee—human last common ancestor Gorilla—human last common ancestor Orangutan—human last common ancestor Gibbon —human last common ancestor List of fictional primates non-human Great apes Human evolution Monkey Day Mythic humanoids Sasquatch Yeren Yeti Yowie. Thomas Brenna, PhD". I think that we need to formulate a new overall-theory, a new anthropological paradigm, about the origin of man. This idea has been flourishing since Charles Darwin and I think that many scientists and laymen will have difficulties in accepting the Aquatic Ape Hypothesis — as they believe in our brain rather than in our physical characteristics. Last common ancestors Chimpanzee—human Gorilla—human Orangutan—human Gibbon—human. I can see two possible future scenarios for the Aquatic Ape Theory. University The Aquatic Ape Hypothesis: Most Credible Theory of Human Evolution Chicago Press. Human Origins Retrieved 16 January The AAH is generally ignored by anthropologists, although it has a following outside academia and has received celebrity endorsement, for example from David Attenborough. -

Looking for Bigfoot LEVELED BOOK • O a Reading A–Z Level O Leveled Book Word Count: 714 Looking for Bigfoot
Looking for Bigfoot LEVELED BOOK • O A Reading A–Z Level O Leveled Book Word Count: 714 Looking for Bigfoot Written by Torran Anderson Illustrated by Norm Grock Visit www.readinga-z.com www.readinga-z.com for thousands of books and materials. Photo Credits: Page 2: © iStockphoto.com/JLF Capture; page 6: © Design Pics Inc./Alamy; page 8: © REUTERS; page 10: © Jesse Harlan Alderman/AP Images; page 11: © Anthony Robert La Penna/Bangor Daily News/The Image Works; page 12: Looking for © Topham/Fortean/The Image Works Bigfoot Looking for Bigfoot Written by Torran Anderson Level O Leveled Book © Learning A–Z Correlation Illustrated by Norm Grock Written by Torran Anderson LEVEL O Illustrated by Norm Grock Fountas & Pinnell M All rights reserved. Reading Recovery 20 DRA 28 www.readinga-z.com www.readinga-z.com Bigfoot Around the World I Set a Trap! Bigfoot or Sasquatch Almas in the Yeti or Migoi in Hibagon in the United States Caucasus Mountains the Himalayas in Japan No one has ever caught Bigfoot before. and Canada Some people think the giant hairy creatures don’t even exist, but I think that Bigfoot is real. To prove it, I’m going to catch one tonight! Then I’m going to take it to school as my science fair project. I can’t wait to see the look on the other kids’ faces when I come to class with Bigfoot. I’m guaranteed to get first place in the science fair. Mapinguari Kikomba Orang Pendek Yowie in Brazil in Africa in Sumatra in Australia Table of Contents I Set a Trap! ............................................... -

The Changing Peasant: Part 2: the Uprooted
19791No. 41 by Richard Critchfield The Changing Peasant Asia [ RC-4-'793 Part II: The Uprooted Theirs is the true lost horizon. Whatever their number, most jutting rocks of shale, schist, and Tibetans are nomadic shepherds limestone. Tibet is the source of all The Tibetan homeland is still so who live in yak-hair tents and move the great rivers of Asia: the Sutlej remote, unmapped, unsurveyed, about much of the year in search of and Indus, the Ganges and and unexplored that in 1979 less is grass for their large herds of yaks, Brahmaputra, the Salween, known about its great Chang Tang sheep, goats, and horses. Aside Yangtze, and Mekong. Violent plateau and Himalayan-high from Lhasa (population 50,000) and gales, dust storms, and icy winds mountain ranges than is known some town-like monasteries, before blow almost every day. about the craters of the moon. the Chinese occupation almost no Until the Chinese occupation the human habitations existed; the Tibet remains a mystery. wheel, considered sacred, was Tibetan herdsman's life, like that of never used: horses, camels, yaks, the Arab Bedouin, revolves around sheep, and men provided the only It is now 29 years since Mao Tse- his livestock, their wool for clothing tung's armies invaded what had transport over rugged tracks and and shelter and their meat and milk precarious bridges spanning deep almost always been an independent for food. sovereign country and declared it an narrow gorges. Even the way of autonomous area within the The Tibetans are racially and death was harsh, the bodies being People's Republic of China. -

TITLE Abstractu, the System Include a Description,Of. The
DOCUMENT RESUME Parmenter, Trevor R. TITLE Developing Independence TfiroUgh Work Prep ration_ Minerma Street Special_ School Augmanted Evaluation of Innovations Program Project `(76/644©) SPONS AGENCY Australian..SchoolsCommission, canberra. PUB DATE 78 NOTE 342p.; The preparation of thiS report wa supparte by the New South Wales State/ Evaluation;Sub- committe of the Australian-Schools ComMission- 'FDPS PRICE MF01/PC14 Plus Postage. DESCRIPTORS_ Conceptual Schemes; *Educabrs Men_ lly-Handiiappe Evaluation Methods; Foreign Countrie_s;Men4:atly. Handicapped-; Models; *Systems Approach; *Work Experience Programs -IDENTIFIERS- *Australia ABSTRACTu, 'Using a systems model ,with its componerits,of inputs -process, atel,witputs; the report evaluates a work eitperiencpprogram formildlyretaded.students at an-Australian special school, Data;,,, -colleted at each Stage of the 'evaluation is presented. Inputs into the system include a_description,of. the population and:itsteeds,, Along with the situational variables that impinge. upon jt...These include the characteristics and value systems of the S-chool -and -i community- and the current economic climate. The aims. and objectives of the-'program-Hare .outlined4 together .with the special programO, techniques, and resources which. were applied. These cover such areas-. as reading, mathematics, Occupationaltherapy,,indastrial arts,- social development, science, and language development-. The dynamics of the organization cif-the proCeSs -variables' are described. -Objecttve assesSment,both criterion-referenced and normative, of the.prograti outcomes are made in each of thecomponentareas. Ratings of the effectiveness of the program -by teachers, parents, students, and. employers are analyzed, along with the predictive value of vocational guidance-tests administered -to each student prior to -the Commencement of the program. (DDS) 4****** Reproductionssupplied by FDRS are the best that can be.made *- * from the' original document.' *** *** ******************************** ** * **** ** VI IV DEPARTNiA T,OFHEALT14. -

With Rex Gilroy
with REX GILROY N.Z Paranormalist, Mark Wallbank chats to the Grandfather of Yowie research in Australia The fossilised Tyrannosaurid footprint discovered by Rex Gilroy with casts of some of the many Yowie Rex Gilroy, Director of the Australian Yowie Research Rex Gilroy in Sydney’s Kuringai National Park on footprints from his collection, together with others from Centre, Katoomba, NSW studying the Wadbilliga Rex Gilroy, Director of the Australian Yowie Research you remember the moment when all the great discoveries I have made in Wednesday 5th May, 2010. When it was first discovered South-east Asia, China, Russia and North America. Dryopithecine fossil footprint. It is yet one more piece of Centre, Katoomba, NSW studying the Wadbilliga the yowie spark ignited and your a lifetime’s research. I have gathered the filled with leaf litter. Photo copyright © Rex Gilroy 2011 Photo copyright © Rex Gilroy 2013. evidence of an Australian primate presence in Pleistocene Dryopithecine fossil footprint. It is yet one more piece of obsession started? largest privately owned natural science times and earlier. Photo copyright © Rex Gilroy 2013. evidence of an Australian primate presence in Pleistocene I grew up on a farm on the Georges River collection in Australia, in the course times and earlier. Photo copyright © Rex Gilroy 2013. of the marsupial hide garments they wore/ fraternity. No-one can say that I have not Moa, three UFO books among others to at Lansvale, outside Cabramatta [born of which I have made many important wear. Being Homo erectus they also know carried out my research and gathered be put on disc and off to the printers in first heard about Rex Gilroy as Nov 8th 1943]. -

Australian Aboriginal Verse 179 Viii Black Words White Page
Australia’s Fourth World Literature i BLACK WORDS WHITE PAGE ABORIGINAL LITERATURE 1929–1988 Australia’s Fourth World Literature iii BLACK WORDS WHITE PAGE ABORIGINAL LITERATURE 1929–1988 Adam Shoemaker THE AUSTRALIAN NATIONAL UNIVERSITY E PRESS iv Black Words White Page E PRESS Published by ANU E Press The Australian National University Canberra ACT 0200, Australia Email: [email protected] Web: http://epress.anu.edu.au Previously published by University of Queensland Press Box 42, St Lucia, Queensland 4067, Australia National Library of Australia Cataloguing-in-Publication entry Black Words White Page Shoemaker, Adam, 1957- . Black words white page: Aboriginal literature 1929–1988. New ed. Bibliography. Includes index. ISBN 0 9751229 5 9 ISBN 0 9751229 6 7 (Online) 1. Australian literature – Aboriginal authors – History and criticism. 2. Australian literature – 20th century – History and criticism. I. Title. A820.989915 All rights reserved. You may download, display, print and reproduce this material in unaltered form only (retaining this notice) for your personal, non-commercial use or use within your organization. All electronic versions prepared by UIN, Melbourne Cover design by Brendon McKinley with an illustration by William Sandy, Emu Dreaming at Kanpi, 1989, acrylic on canvas, 122 x 117 cm. The Australian National University Art Collection First edition © 1989 Adam Shoemaker Second edition © 1992 Adam Shoemaker This edition © 2004 Adam Shoemaker Australia’s Fourth World Literature v To Johanna Dykgraaf, for her time and care -

Research Article WILDMEN in MYANMAR
The RELICT HOMINOID INQUIRY 4:53-66 (2015) Research Article WILDMEN IN MYANMAR: A COMPENDIUM OF PUBLISHED ACCOUNTS AND REVIEW OF THE EVIDENCE Steven G. Platt1, Thomas R. Rainwater2* 1 Wildlife Conservation Society-Myanmar Program, Office Block C-1, Aye Yeik Mon 1st Street, Hlaing Township, Yangon, Myanmar 2 Baruch Institute of Coastal Ecology and Forest Science, Clemson University, P.O. Box 596, Georgetown, SC 29442, USA ABSTRACT. In contrast to other countries in Asia, little is known concerning the possible occurrence of undescribed Hominoidea (i.e., wildmen) in Myanmar (Burma). We here present six accounts from Myanmar describing wildmen or their sign published between 1910 and 1972; three of these reports antedate popularization of wildmen (e.g., yeti and sasquatch) in the global media. Most reports emanate from mountainous regions of northern Myanmar (primarily Kachin State) where wildmen appear to inhabit montane forests. Wildman tracks are described as superficially similar to human (Homo sapiens) footprints, and about the same size to almost twice the size of human tracks. Presumptive pressure ridges were described in one set of wildman tracks. Accounts suggest wildmen are bipedal, 120-245 cm in height, and covered in longish pale to orange-red hair with a head-neck ruff. Wildmen are said to utter distinctive vocalizations, emit strong odors, and sometimes behave aggressively towards humans. Published accounts of wildmen in Myanmar are largely based on narratives provided by indigenous informants. We found nothing to indicate informants were attempting to beguile investigators, and consider it unlikely that wildmen might be confused with other large mammals native to the region. -
Mythical Creatures
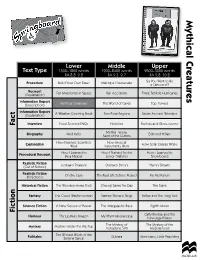
Mythical Creatures Lower Middle Upper Text Type 1500–1800 words 1900–2400 words 2500–3000 words RA 8.8–9.2 RA 9.3–9.7 RA 9.8–10.2 So You Want to Be Procedure Build Your Own Easel Making a Cheesecake a Cartoonist? Recount (Explanation) Ten Milestones in Space Rail Accidents Three Terrible Hurricanes Information Report (Description) Mythical Creatures The World of Caves Top Towers Information Report (Explanation) A Weather Counting Book Two Polar Regions Seven Ancient Wonders Fact Interview Food Science FAQs Hobbies Fireflies and Glow-worms Mother Teresa: Biography Ned Kelly Saint of the Gutters Edmund Hillary How Forensic Scientists How Musical Explanation Work Instruments Work How Solar Energy Works How I Learned to How I Trained for the How I Learned to Procedural Recount Be a Nipper Junior Triathlon Snowboard Realistic Fiction (Out of School) Junkyard Treasure Outback Betty’s Harry’s Dream Realistic Fiction (In School) On the Case The Real-Life School Project Ms McMahon Historical Fiction The Wooden Horse Trick Cheung Saves the Day The Slave Fantasy The Cloud Washerwoman Sammy Stevens Sings Finbar and the Long Trek Science Fiction A New Source of Power The Intergalactic Race Eighth Moon Fiction Catty Bimbar and the Humour The Upstairs Dragon My Rhyming Grandpa New-Age Pirates The Mystery of The Mystery of the Mystery Mystery Under the Big Top Autoplane 500 Missing Food The Wicked Witch of the Folktales Singing Sands Gulnara Momotaro, Little Peachling We have designed these lesson plans so that you can have the plan in front of you as you teach, along with a copy of the book. -

The Friendly Yeti
The University of Southern Mississippi The Aquila Digital Community Faculty Publications 2012 The Friendly Yeti Daniel S. Capper University of Southern Mississippi, [email protected] Follow this and additional works at: https://aquila.usm.edu/fac_pubs Part of the Animal Studies Commons, Buddhist Studies Commons, and the South and Southeast Asian Languages and Societies Commons Recommended Citation Capper, D. S. (2012). The Friendly Yeti. Journal for the Study of Religion, Nature, and Culture, 6(1), 71-87. Available at: https://aquila.usm.edu/fac_pubs/14855 This Article is brought to you for free and open access by The Aquila Digital Community. It has been accepted for inclusion in Faculty Publications by an authorized administrator of The Aquila Digital Community. For more information, please contact [email protected]. This article appeared in the Journal for the Study of Religion, Nature, and Culture 6:1 (2012): 71-87. THE FRIENDLY YETI Daniel Capper, Ph.D. Associate Professor of Religion The University of Southern Mississippi 118 College Drive, #5015 Hattiesburg, MS 39406 USA 601-266-4522 [email protected] ABSTRACT Most images of yetis in Western popular culture and scholarly literature portray them as secular, predatory monsters. These representations overlook important religious dimensions of yetis that are hidden in the current literature so I take a new look at yetis in Tibetan religions in order to clarify our understanding of these legendary creatures. Following a phenomenological approach that sets aside the issue of the ontological existence of yetis I examine texts, art, ritual, and folklore in order to propose four yeti personal ideal types: the Buddhist practitioner, the human religious ally, the friendly yeti, and the mountain deity yeti.