Mutation Screen of the SIM1 Gene in Pediatric Patients with Early-Onset Obesity
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

1 Evidence for Gliadin Antibodies As Causative Agents in Schizophrenia
1 Evidence for gliadin antibodies as causative agents in schizophrenia. C.J.Carter PolygenicPathways, 20 Upper Maze Hill, Saint-Leonard’s on Sea, East Sussex, TN37 0LG [email protected] Tel: 0044 (0)1424 422201 I have no fax Abstract Antibodies to gliadin, a component of gluten, have frequently been reported in schizophrenia patients, and in some cases remission has been noted following the instigation of a gluten free diet. Gliadin is a highly immunogenic protein, and B cell epitopes along its entire immunogenic length are homologous to the products of numerous proteins relevant to schizophrenia (p = 0.012 to 3e-25). These include members of the DISC1 interactome, of glutamate, dopamine and neuregulin signalling networks, and of pathways involved in plasticity, dendritic growth or myelination. Antibodies to gliadin are likely to cross react with these key proteins, as has already been observed with synapsin 1 and calreticulin. Gliadin may thus be a causative agent in schizophrenia, under certain genetic and immunological conditions, producing its effects via antibody mediated knockdown of multiple proteins relevant to the disease process. Because of such homology, an autoimmune response may be sustained by the human antigens that resemble gliadin itself, a scenario supported by many reports of immune activation both in the brain and in lymphocytes in schizophrenia. Gluten free diets and removal of such antibodies may be of therapeutic benefit in certain cases of schizophrenia. 2 Introduction A number of studies from China, Norway, and the USA have reported the presence of gliadin antibodies in schizophrenia 1-5. Gliadin is a component of gluten, intolerance to which is implicated in coeliac disease 6. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

User's Manual Mobile Phone
User’s Manual Mobile Phone Model: AM206 FCC ID: UOSAM206 1 TABLE OF CONTENT 1. BASIC INFORMATION .................... 6 1.1. BRIEF INTRODUCTION ........................................... 6 2. BEFORE USE ..................................... 7 2.1. NAME AND EXPLANATION OF EACH PART ............. 7 2.1.1. Appearance Sketch Map ........................... 7 2.1.2. Description of the Standby Pictures .......... 7 2.1.3. Description of Keys .................................. 8 2.2. INSTALL SIM CARD AND MEMORY CARD ........... 10 2.2.1. SIM Card ................................................. 10 2.2.2. Inserting and Taking out SIM Card ........ 11 2.2.3. Memory Card .......................................... 12 2.3. BATTERY ............................................................ 12 2.3.1. Install the Battery .................................... 12 2.3.2. Charging .................................................. 13 2.4. SECURITY PASSWORD ......................................... 14 2 3. QUICK USER GUIDE ..................... 15 3.1. DIAL A CALL ....................................................... 15 3.2. REJECT A CALL ................................................... 15 3.3. RECEIVE A CALL ................................................. 15 3.4. END A CALL ........................................................ 16 3.5. EMERGENCY SERVICES ....................................... 16 3.6. EXTENSION SPEED DIAL ..................................... 16 3.7. DIALED CALLS/MISSED CALLS/RECEIVED CALLS/REJECTED CALLS ............................................. -

Supplementary Materials
Supplementary materials Supplementary Table S1: MGNC compound library Ingredien Molecule Caco- Mol ID MW AlogP OB (%) BBB DL FASA- HL t Name Name 2 shengdi MOL012254 campesterol 400.8 7.63 37.58 1.34 0.98 0.7 0.21 20.2 shengdi MOL000519 coniferin 314.4 3.16 31.11 0.42 -0.2 0.3 0.27 74.6 beta- shengdi MOL000359 414.8 8.08 36.91 1.32 0.99 0.8 0.23 20.2 sitosterol pachymic shengdi MOL000289 528.9 6.54 33.63 0.1 -0.6 0.8 0 9.27 acid Poricoic acid shengdi MOL000291 484.7 5.64 30.52 -0.08 -0.9 0.8 0 8.67 B Chrysanthem shengdi MOL004492 585 8.24 38.72 0.51 -1 0.6 0.3 17.5 axanthin 20- shengdi MOL011455 Hexadecano 418.6 1.91 32.7 -0.24 -0.4 0.7 0.29 104 ylingenol huanglian MOL001454 berberine 336.4 3.45 36.86 1.24 0.57 0.8 0.19 6.57 huanglian MOL013352 Obacunone 454.6 2.68 43.29 0.01 -0.4 0.8 0.31 -13 huanglian MOL002894 berberrubine 322.4 3.2 35.74 1.07 0.17 0.7 0.24 6.46 huanglian MOL002897 epiberberine 336.4 3.45 43.09 1.17 0.4 0.8 0.19 6.1 huanglian MOL002903 (R)-Canadine 339.4 3.4 55.37 1.04 0.57 0.8 0.2 6.41 huanglian MOL002904 Berlambine 351.4 2.49 36.68 0.97 0.17 0.8 0.28 7.33 Corchorosid huanglian MOL002907 404.6 1.34 105 -0.91 -1.3 0.8 0.29 6.68 e A_qt Magnogrand huanglian MOL000622 266.4 1.18 63.71 0.02 -0.2 0.2 0.3 3.17 iolide huanglian MOL000762 Palmidin A 510.5 4.52 35.36 -0.38 -1.5 0.7 0.39 33.2 huanglian MOL000785 palmatine 352.4 3.65 64.6 1.33 0.37 0.7 0.13 2.25 huanglian MOL000098 quercetin 302.3 1.5 46.43 0.05 -0.8 0.3 0.38 14.4 huanglian MOL001458 coptisine 320.3 3.25 30.67 1.21 0.32 0.9 0.26 9.33 huanglian MOL002668 Worenine -

Brain Nuclear Receptors and Body Weight Regulation
Brain nuclear receptors and body weight regulation Yong Xu, … , Bert W. O’Malley, Joel K. Elmquist J Clin Invest. 2017;127(4):1172-1180. https://doi.org/10.1172/JCI88891. Review Series Neural pathways, especially those in the hypothalamus, integrate multiple nutritional, hormonal, and neural signals, resulting in the coordinated control of body weight balance and glucose homeostasis. Nuclear receptors (NRs) sense changing levels of nutrients and hormones, and therefore play essential roles in the regulation of energy homeostasis. Understanding the role and the underlying mechanisms of NRs in the context of energy balance control may facilitate the identification of novel targets to treat obesity. Notably, NRs are abundantly expressed in the brain, and emerging evidence indicates that a number of these brain NRs regulate multiple aspects of energy balance, including feeding, energy expenditure and physical activity. In this Review we summarize some of the recent literature regarding effects of brain NRs on body weight regulation and discuss mechanisms underlying these effects. Find the latest version: http://jci.me/88891/pdf REVIEW SERIES: NUCLEAR RECEPTORS The Journal of Clinical Investigation Series Editor: Mitchell A. Lazar Brain nuclear receptors and body weight regulation Yong Xu,1,2 Bert W. O’Malley,2 and Joel K. Elmquist3 1Children’s Nutrition Research Center, Department of Pediatrics, and 2Department of Molecular and Cellular Biology, Baylor College of Medicine, Houston, Texas, USA. 3Division of Hypothalamic Research, Department of Internal Medicine, University of Texas Southwestern Medical Center, Dallas, Texas, USA. Neural pathways, especially those in the hypothalamus, integrate multiple nutritional, hormonal, and neural signals, resulting in the coordinated control of body weight balance and glucose homeostasis. -
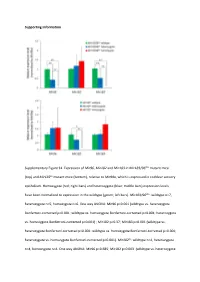
Supporting Information Supplementary Figure S1
Supporting Information Supplementary Figure S1. Expression of Mir96, Mir182 and Mir183 in Mir183/96dko mutant mice (top) and Mir182ko mutant mice (bottom), relative to Mir99a, which is expressed in cochlear sensory epithelium. Homozygote (red; right bars) and heterozygote (blue; middle bars) expression levels have been normalised to expression in the wildtype (green; left bars). Mir183/96dko: wildtype n=7, heterozygote n=5, homozygote n=6. One way ANOVA: Mir96 p<0.001 (wildtype vs. heterozygote Bonferroni‐corrected p<0.001; wildtype vs. homozygote Bonferroni‐corrected p<0.001; heterozygote vs. homozygote Bonferroni‐corrected p=0.001) ; Mir182 p=0.37; Mir183 p<0.001 (wildtype vs. heterozygote Bonferroni‐corrected p=0.001; wildtype vs. homozygote Bonferroni‐corrected p<0.001; heterozygote vs. homozygote Bonferroni‐corrected p<0.001). Mir182ko: wildtype n=4, heterozygote n=4, homozygote n=4. One way ANOVA: Mir96 p=0.685; Mir182 p=0.003 (wildtype vs. heterozygote Bonferroni‐corrected p=0.397; wildtype vs. homozygote Bonferroni‐corrected p=0.003; heterozygote vs. homozygote Bonferroni‐corrected p=0.032); Mir183 p=0.04 (wildtype vs. heterozygote Bonferroni‐corrected p=1.0; wildtype vs. homozygote Bonferroni‐corrected p=0.068; heterozygote vs. homozygote Bonferroni‐corrected p=0.094), Error bars are standard deviation (* = P < 0.05, ** = P ≤ 0.01). Supplementary Figure S2. Individual ABR thresholds of wildtype, heterozygous and homozygous Mir183/96dko mice at all ages tested. Number of mice of each genotype tested at each age is shown on the threshold plot. Supplementary Figure S3. Individual ABR thresholds of wildtype, heterozygous and homozygous Mir182ko mice at all ages tested. -
Reprogramming of Lysosomal Gene Expression by Interleukin-4 and Stat6 Brignull Et Al
Reprogramming of lysosomal gene expression by interleukin-4 and Stat6 Brignull et al. Brignull et al. BMC Genomics 2013, 14:853 http://www.biomedcentral.com/1471-2164/14/853 Brignull et al. BMC Genomics 2013, 14:853 http://www.biomedcentral.com/1471-2164/14/853 RESEARCH ARTICLE Open Access Reprogramming of lysosomal gene expression by interleukin-4 and Stat6 Louise M Brignull1†, Zsolt Czimmerer2†, Hafida Saidi1,3, Bence Daniel2, Izabel Villela4,5, Nathan W Bartlett6, Sebastian L Johnston6, Lisiane B Meira4, Laszlo Nagy2,7 and Axel Nohturfft1* Abstract Background: Lysosomes play important roles in multiple aspects of physiology, but the problem of how the transcription of lysosomal genes is coordinated remains incompletely understood. The goal of this study was to illuminate the physiological contexts in which lysosomal genes are coordinately regulated and to identify transcription factors involved in this control. Results: As transcription factors and their target genes are often co-regulated, we performed meta-analyses of array-based expression data to identify regulators whose mRNA profiles are highly correlated with those of a core set of lysosomal genes. Among the ~50 transcription factors that rank highest by this measure, 65% are involved in differentiation or development, and 22% have been implicated in interferon signaling. The most strongly correlated candidate was Stat6, a factor commonly activated by interleukin-4 (IL-4) or IL-13. Publicly available chromatin immunoprecipitation (ChIP) data from alternatively activated mouse macrophages show that lysosomal genes are overrepresented among Stat6-bound targets. Quantification of RNA from wild-type and Stat6-deficient cells indicates that Stat6 promotes the expression of over 100 lysosomal genes, including hydrolases, subunits of the vacuolar H+ ATPase and trafficking factors. -

Transcription Factor -Catenin Plays a Key Role in Fluid Flow Shear Stress
cells Article Transcription Factor β-Catenin Plays a Key Role in Fluid Flow Shear Stress-Mediated Glomerular Injury in Solitary Kidney Tarak Srivastava 1,2,3,*, Daniel P. Heruth 4 , R. Scott Duncan 5, Mohammad H. Rezaiekhaligh 1, Robert E. Garola 6, Lakshmi Priya 1, Jianping Zhou 2,7, Varun C. Boinpelly 2,7, Jan Novak 8, Mohammed Farhan Ali 1, Trupti Joshi 9,10,11,12, Uri S. Alon 1, Yuexu Jiang 10,11, Ellen T. McCarthy 13, Virginia J. Savin 7, Ram Sharma 7, Mark L. Johnson 3 and Mukut Sharma 2,7,13 1 Section of Nephrology, Children’s Mercy Hospital and University of Missouri at Kansas City, Kansas City, MO 64108, USA; [email protected] (M.H.R.); [email protected] (L.P.); [email protected] (M.F.A.); [email protected] (U.S.A.) 2 Midwest Veterans’ Biomedical Research Foundation (MVBRF), Kansas City, MO 64128, USA; [email protected] (J.Z.); [email protected] (V.C.B.); [email protected] (M.S.) 3 Department of Oral and Craniofacial Sciences, School of Dentistry, University of Missouri at Kansas City, Kansas City, MO 64108, USA; [email protected] 4 Children’s Mercy Research Institute, Children’s Mercy Hospital and University of Missouri at Kansas City, Kansas City, MO 64108, USA; [email protected] 5 School of Biological Sciences, University of Missouri at Kansas City, Kansas City, MO 64108, USA; [email protected] 6 Department of Pathology and Laboratory Medicine, Children’s Mercy Hospital and University of Missouri at Kansas City, Kansas City, MO 64108, USA; [email protected] 7 Kansas City VA Medical Center, Kansas City, MO 64128, USA; [email protected] (V.J.S.); Citation: Srivastava, T.; Heruth, D.P.; [email protected] (R.S.) 8 Duncan, R.S.; Rezaiekhaligh, M.H.; Department of Microbiology, University of Alabama at Birmingham, Birmingham, AL 35487, USA; Garola, R.E.; Priya, L.; Zhou, J.; [email protected] 9 Department of Health Management and Informatics, University of Missouri, Columbia, MO 65211, USA; Boinpelly, V.C.; Novak, J.; Ali, M.F.; [email protected] et al. -

Deletion of the Sclerotome-Enriched Lncrna PEAT Augments Ribosomal Protein Expression
Deletion of the sclerotome-enriched lncRNA PEAT augments ribosomal protein expression David A. Stafforda, Darwin S. Dichmanna, Jessica K. Changb, and Richard M. Harlanda,1 aDepartment of Molecular and Cell Biology, University of California, Berkeley, CA 94720; and bDepartment of Genetics, Stanford University, Stanford, CA 94305-5120 Contributed by Richard M. Harland, September 23, 2016 (sent for review July 22, 2016; reviewed by Margaret Buckingham and Chen-Ming Fan) To define a complete catalog of the genes that are activated during developing sclerotome by dissection from early embryos at 8- to mouse sclerotome formation, we sequenced RNA from embryonic 9.5-d postconception is technically difficult. Although a model mouse tissue directed to form sclerotome in culture. In addition to for presomitic mesoderm culture from stem cells has been de- well-known early markers of sclerotome, such as Pax1, Pax9,andthe veloped (10), this has not been exploited as an in vitro model of Bapx2/Nkx3-2 homolog Nkx3-1, the long-noncoding RNA PEAT (Pax1 the sclerotome. To obtain sufficient sclerotomal transcripts for enhancer antisense transcript) was induced in sclerotome-directed RNA-seq, we chemically treated explants of embryonic tissue to samples. Strikingly, PEAT is located just upstream of the Pax1 gene. bias differentiation of fluorescently marked presomitic mesoderm Using CRISPR/Cas9, we generated a mouse line bearing a complete toward the sclerotome. Following FACS, we compared control deletion of the PEAT-transcribed unit. RNA-seq on PEAT mutant and induced cultures to define the sclerotome transcriptome. In embryos showed that loss of PEAT modestly increases bone mor- addition to the previously described transcription factors, we identi- phogenetic protein target gene expression and also elevates the fied an uncharacterized noncoding RNA transcript that we named expression of a large subset of ribosomal protein mRNAs. -

The Pennsylvania State University the Graduate School College Of
The Pennsylvania State University The Graduate School College of Agricultural Sciences GENOMIC AND IMMUNOLOGICAL APPROACHES TO CONTROL GRAM- NEGATIVE INTRAMAMMARY INFECTIONS IN DAIRY CATTLE A Thesis in Animal Science by Annapoorani Chockalingam © 2006 Annapoorani Chockalingam Submitted in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy May 2006 The thesis of Annapoorani Chockalingam was reviewed and approved* by the following: Cindy E. McKinney Assistant Professor of Transgenic Biology Thesis Advisor Co-Chair of Committee Douglas D. Bannerman Research Scientist, BFGL-ANRI, BARC, USDA-ARS Co-Chair of Committee Guy F. Barbato Associate Professor of Poultry Science Kenneth M. Weiss Evan Pugh Professor of Anthropology and Genetics Eric T. Harvill Assistant Professor of Veterinary Science Robert F. Paulson Associate Professor of Veterinary Science Terry D. Etherton Distinguished Professor of Animal Nutrition Head of the Department of Dairy and Animal Science *Signatures are on file in the Graduate School iii ABSTRACT The incidence of mastitis due to Gram-negative bacteria continues to impact the dairy industry due to the ubiquitous nature of the pathogens. Gram-negative bacteria account for nearly 40% of clinical mastitis cases resulting in an estimated loss to the U.S. dairy industry of $800 million per year. The research presented here focuses on host- mediated genetic and immunological factors involved in Gram-negative bacteria induced mastitis during the early phases of disease onset. Array analysis of mammary gland gene expression in a global context in response to E. coli is currently unavailable. A mouse mastitis model was used to study the transcriptional response in the mammary gland. -

Metabolic and Behavioral Effects of Mutant Huntingtin Deletion in Sim1
www.nature.com/scientificreports OPEN Metabolic and behavioral effects of mutant huntingtin deletion in Sim1 neurons in the BACHD mouse Received: 08 April 2016 Accepted: 31 May 2016 model of Huntington’s disease Published: 23 June 2016 Rana Soylu-Kucharz, Barbara Baldo & Åsa Petersén Hypothalamic pathology, metabolic dysfunction and psychiatric symptoms are part of Huntington disease (HD), which is caused by an expanded CAG repeat in the huntingtin (HTT) gene. Inactivation of mutant HTT selectively in the hypothalamus prevents the development of metabolic dysfunction and depressive-like behavior in the BACHD mouse model. The hypothalamic paraventricular nucleus (PVN) is implicated in metabolic and emotional control, therefore we here tested whether inactivation of mutant HTT in the PVN affects metabolic and psychiatric manifestations of HD in BACHD mice. BACHD mice were crossed with mice expressing Cre-recombinase under the Sim1 promoter (Sim1- Cre) to inactivate mutant HTT in Sim1 expressing cells, i.e. the PVN of the hypothalamus. We found that inactivation of mutant HTT in Sim1 cells had a sex-specific effect on both the metabolic and the psychiatric phenotype, as these phenotypes were no longer different in male BACHD/Sim1-Cre mice compared to wild-type littermates. We also found a reduced number of GnRH neurons specifically in the anterior hypothalamus and an increased testes weight in male BACHD mice compared to wild- type littermates. Taken together, expression of mutant HTT in Sim1 cells may play a role for the development of metabolic dysfunction and depressive-like behavior in male BACHD mice. Huntington’s disease (HD) is a neurodegenerative disorder with formal diagnosis based on prominent motor symptoms and a positive gene test for an expansion of the CAG repeat in the huntingtin (HTT) gene1,2. -

Ventral Neural Patterning by Nkx Homeobox Genes: Nkx6.1 Controls Somatic Motor Neuron and Ventral Interneuron Fates
Downloaded from genesdev.cshlp.org on September 30, 2021 - Published by Cold Spring Harbor Laboratory Press RESEARCH COMMUNICATION vitro in response to two- to threefold changes in the Ventral neural patterning by concentration of Shh, and the position at which each Nkx homeobox genes: Nkx6.1 neuronal class is generated in vivo is predicted by the controls somatic motor neuron concentration required for their induction in vivo (Eric- son et al. 1997a,b; Briscoe et al. 2000). Thus, neurons and ventral interneuron fates generated in more ventral regions of the neural tube re- 1,2,6 3,6 quire progressively higher concentrations of Shh for their Maike Sander, Sussan Paydar, induction. 4,5 4 Johan Ericson, James Briscoe, The genetic programs activated in neural progenitor 3 1 Elizabeth Berber, Michael German, cells in response to Shh signaling, however, remain in- Thomas M. Jessell,4 and John L.R. Rubenstein3,7 completely defined. Emerging evidence suggests that homeobox genes function as critical intermediaries in 1Hormone Research Institute, Department of Medicine, the neural response to Shh signals (Lumsden and Krum- University of California–San Francisco, San Franscisco, California 94143 USA; 2Center for Molecular Neurobiology, lauf 1996; Tanabe and Jessell 1996; Ericson et al. 1997a,b; Hamburg, Germany; 3Nina Ireland Laboratory of Hammerschmidt et al. 1997; Rubenstein et al. 1998). Developmental Neurobiology, University of California–San Several homeobox genes are expressed by ventral pro- Francisco, San Francisco, California 94143 USA;