An Improved Process for the Preparation of Pure Cyamemazine and Salts Thereof
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Information for the Patient Losartan Potassium/Hydrochlorothiazide 100
Package leaflet: Information for the patient Losartan Potassium/Hydrochlorothiazide 100 mg/12.5 mg Film-coated Tablets (losartan potassium/hydrochlorothiazide) Read all of this leaflet carefully before you start taking this medicine because it contains important information for you. - Keep this leaflet. You may need to read it again. - If you have any further questions, ask your doctor or pharmacist. - This medicine has been prescribed for you only. Do not pass it on to others. It may harm them, even if their signs of illness are the same as yours. - If you get any side effects, talk to your doctor or pharmacist. This includes any possible side effects not listed in this leaflet. See section 4. What is in this leaflet 1. What Losartan Potassium/Hydrochlorothiazide is and what it is used for 2. What you need to know before you take Losartan Potassium/Hydrochlorothiazide 3. How to take Losartan Potassium/Hydrochlorothiazide 4. Possible side effects 5. How to store Losartan Potassium/Hydrochlorothiazide 6. Contents of the pack and other information 1. What Losartan Potassium/Hydrochlorothiazide is and what it is used for Losartan Potassium/Hydrochlorothiazide is a combination of an angiotensin II receptor antagonist (losartan) and a diuretic (hydrochlorothiazide). Angiotensin II is a substance produced in the body which binds to receptors in blood vessels, causing them to tighten. This results in an increase in blood pressure. Losartan prevents the binding of angiotensin II to these receptors, causing the blood vessels to relax which in turn lowers the blood pressure. Hydrochlorothiazide works by making the kidneys pass more water and salt. -

The Effects of Antipsychotic Treatment on Metabolic Function: a Systematic Review and Network Meta-Analysis
The effects of antipsychotic treatment on metabolic function: a systematic review and network meta-analysis Toby Pillinger, Robert McCutcheon, Luke Vano, Katherine Beck, Guy Hindley, Atheeshaan Arumuham, Yuya Mizuno, Sridhar Natesan, Orestis Efthimiou, Andrea Cipriani, Oliver Howes ****PROTOCOL**** Review questions 1. What is the magnitude of metabolic dysregulation (defined as alterations in fasting glucose, total cholesterol, low density lipoprotein (LDL) cholesterol, high density lipoprotein (HDL) cholesterol, and triglyceride levels) and alterations in body weight and body mass index associated with short-term (‘acute’) antipsychotic treatment in individuals with schizophrenia? 2. Does baseline physiology (e.g. body weight) and demographics (e.g. age) of patients predict magnitude of antipsychotic-associated metabolic dysregulation? 3. Are alterations in metabolic parameters over time associated with alterations in degree of psychopathology? 1 Searches We plan to search EMBASE, PsycINFO, and MEDLINE from inception using the following terms: 1 (Acepromazine or Acetophenazine or Amisulpride or Aripiprazole or Asenapine or Benperidol or Blonanserin or Bromperidol or Butaperazine or Carpipramine or Chlorproethazine or Chlorpromazine or Chlorprothixene or Clocapramine or Clopenthixol or Clopentixol or Clothiapine or Clotiapine or Clozapine or Cyamemazine or Cyamepromazine or Dixyrazine or Droperidol or Fluanisone or Flupehenazine or Flupenthixol or Flupentixol or Fluphenazine or Fluspirilen or Fluspirilene or Haloperidol or Iloperidone -

Antipsychotics
The Fut ure of Antipsychotic Therapy (page 7 in syllabus) Stepp,,hen M. Stahl, MD, PhD Adjunct Professor, Department of Psychiatry Universityyg of California, San Diego School of Medicine Honorary Visiting Senior Fellow, Cambridge University, UK Sppyonsored by the Neuroscience Education Institute Additionally sponsored by the American Society for the Advancement of Pharmacotherapy This activity is supported by an educational grant from Sunovion Pharmaceuticals Inc. Copyright © 2011 Neuroscience Education Institute. All rights reserved. Individual Disclosure Statement Faculty Editor / Presenter Stephen M. Stahl, MD, PhD, is an adjunct professor in the department of psychiatry at the University of California, San Diego School of Medicine, and an honorary visiting senior fellow at the University of Cambridge in the UK. Grant/Research: AstraZeneca, BioMarin, Dainippon Sumitomo, Dey, Forest, Genomind, Lilly, Merck, Pamlab, Pfizer, PGxHealth/Trovis, Schering-Plough, Sepracor/Sunovion, Servier, Shire, Torrent Consultant/Advisor: Advent, Alkermes, Arena, AstraZeneca, AVANIR, BioMarin, Biovail, Boehringer Ingelheim, Bristol-Myers Squibb, CeNeRx, Cypress, Dainippon Sumitomo, Dey, Forest, Genomind, Janssen, Jazz, Labopharm, Lilly, Lundbeck, Merck, Neuronetics, Novartis, Ono, Orexigen, Otsuka, Pamlab, Pfizer, PGxHealth/Trovis, Rexahn, Roche, Royalty, Schering-Plough, Servier, Shire, Solvay/Abbott, Sunovion/Sepracor, Valeant, VIVUS, Speakers Bureau: Dainippon Sumitomo, Forest, Lilly, Merck, Pamlab, Pfizer, Sepracor/Sunovion, Servier, Wyeth Copyright © 2011 Neuroscience Education Institute. All rights reserved. Learninggj Objectives • Differentiate antipsychotic drugs from each other on the basis of their pharmacological mechanisms and their associated therapeutic and side effects • Integrate novel treatment approaches into clinical practice according to best practices guidelines • Identify novel therapeutic options currently being researched for the treatment of schizophrenia Copyright © 2011 Neuroscience Education Institute. -

Drugs with Anticholinergic Properties, Cognitive Decline, and Dementia in an Elderly General Population: the 3-City Study
Drugs with anticholinergic properties, cognitive decline, and dementia in an elderly general population: the 3-city study. Isabelle Carrière, Annie Fourrier-Reglat, Jean-François Dartigues, Olivier Rouaud, Florence Pasquier, Karen Ritchie, Marie-Laure Ancelin To cite this version: Isabelle Carrière, Annie Fourrier-Reglat, Jean-François Dartigues, Olivier Rouaud, Florence Pasquier, et al.. Drugs with anticholinergic properties, cognitive decline, and dementia in an elderly gen- eral population: the 3-city study.: Anticholinergics and cognitive dysfunction. Archives of Internal Medicine -New Series-, American Medical Association, 2009, 169 (14), pp.1317-24. 10.1001/archin- ternmed.2009.229. inserm-00415864 HAL Id: inserm-00415864 https://www.hal.inserm.fr/inserm-00415864 Submitted on 27 Jul 2010 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Archives of Internal Medicine in press Abstract: 248 words Text: 3441 words Number of Tables: 4 Drugs with anticholinergic properties, cognitive decline, and dementia in an elderly general population. The Three-City -

Screening of 300 Drugs in Blood Utilizing Second Generation
Forensic Screening of 300 Drugs in Blood Utilizing Exactive Plus High-Resolution Accurate Mass Spectrometer and ExactFinder Software Kristine Van Natta, Marta Kozak, Xiang He Forensic Toxicology use Only Drugs analyzed Compound Compound Compound Atazanavir Efavirenz Pyrilamine Chlorpropamide Haloperidol Tolbutamide 1-(3-Chlorophenyl)piperazine Des(2-hydroxyethyl)opipramol Pentazocine Atenolol EMDP Quinidine Chlorprothixene Hydrocodone Tramadol 10-hydroxycarbazepine Desalkylflurazepam Perimetazine Atropine Ephedrine Quinine Cilazapril Hydromorphone Trazodone 5-(p-Methylphenyl)-5-phenylhydantoin Desipramine Phenacetin Benperidol Escitalopram Quinupramine Cinchonine Hydroquinine Triazolam 6-Acetylcodeine Desmethylcitalopram Phenazone Benzoylecgonine Esmolol Ranitidine Cinnarizine Hydroxychloroquine Trifluoperazine Bepridil Estazolam Reserpine 6-Monoacetylmorphine Desmethylcitalopram Phencyclidine Cisapride HydroxyItraconazole Trifluperidol Betaxolol Ethyl Loflazepate Risperidone 7(2,3dihydroxypropyl)Theophylline Desmethylclozapine Phenylbutazone Clenbuterol Hydroxyzine Triflupromazine Bezafibrate Ethylamphetamine Ritonavir 7-Aminoclonazepam Desmethyldoxepin Pholcodine Clobazam Ibogaine Trihexyphenidyl Biperiden Etifoxine Ropivacaine 7-Aminoflunitrazepam Desmethylmirtazapine Pimozide Clofibrate Imatinib Trimeprazine Bisoprolol Etodolac Rufinamide 9-hydroxy-risperidone Desmethylnefopam Pindolol Clomethiazole Imipramine Trimetazidine Bromazepam Felbamate Secobarbital Clomipramine Indalpine Trimethoprim Acepromazine Desmethyltramadol Pipamperone -

Emea/666243/2009
European Medicines Agency London, 29 October 2009 EMEA/666243/2009 ISSUE NUMBER: 0910 MONTHLY REPORT PHARMACOVIGILANCE WORKING PARTY (PHVWP) OCTOBER 2009 PLENARY MEETING The CHMP Pharmacovigilance Working Party (PhVWP) held its October 2009 plenary meeting on 19-21 October 2009. PhVWP DISCUSSIONS ON SAFETY CONCERNS Below is a summary of the discussions regarding non-centrally authorised medicinal products in accordance with the PhVWP publication policy (see under http://www.emea.europa.eu/htms/human/phv/reports.htm). Positions agreed by the PhVWP for non- centrally authorised products are recommendations to Member States. For safety updates concerning centrally authorised products and products subject to ongoing CHMP procedures, readers are referred to the CHMP Monthly Report (see under http://www.emea.europa.eu/pressoffice/presshome.htm). The PhVWP provides advice on these products to the Committee of Medicinal Products for Human Use (CHMP) upon its request. Antipsychotics - risk of venous thromboembolism (VTE) Identify risk factors for VTE for preventive action before and during treatment with antipsychotics The PhVWP completed their review on the risk of VTE of antipsychotics1. The review was triggered by and based on data from the UK spontaneous adverse drug reactions reporting system and the published literature. The PhVWP carefully considered the data, including the limitations of both information sources, such as the lack of randomised controlled trial data, the heterogeneity of published studies and the potential confounding factors such as sedation and weight gain, commonly present in antipsychotic users. The PhVWP concluded that an association between VTE and antipsychotics cannot be excluded. Distinguishing different risk levels between the various active substances was not possible. -

Clozapine/Cyamemazine 985 Verse Effects, Clozapine Therapy Should Be Introduced Account
Clozapine/Cyamemazine 985 verse effects, clozapine therapy should be introduced account. There have been a few reports of exacerbation of psy- Clozapine has shown consistent clinical benefit in schizophrenic chotic symptoms in patients who were switched to a generic for- patients with persistent aggressive or violent behaviour.2,9 gradually, beginning with low doses and increasing ac- 6,7 cording to response. mulation. Whether this is due to a sedative effect, a specific antiaggressive 1. Sajbel TA, et al. Converting patients from brand-name clozapine action, or just reflects an overall improvement in psychosis is un- In the treatment of schizophrenia, including reducing to generic clozapine. Ann Pharmacother 2001; 35: 281–4. known. 2. Makela EH, et al. Branded versus generic clozapine for treat- the risk of suicidal behaviour, the usual oral dose is ment of schizophrenia. Ann Pharmacother 2003; 37: 350–3. Clozapine has been advocated for use in schizophrenic patients 12.5 mg once or twice on the first day followed by 3. Stoner SC, et al. A program to convert patients from trade-name with moderate to severe tardive dyskinesia. It is still unclear 25 mg once or twice on the second day. Thereafter the to generic clozapine. Pharmacotherapy 2003; 23: 806–10. whether clozapine can itself cause tardive dyskinesia but some 4. Bazire S, Burton V. Generic clozapine in schizophrenia: what is patients with established tardive dyskinesia have experienced daily dosage may be increased gradually in steps of 25 all the fuss about? Pharm J 2004; 273: 720–1. improvement in their symptoms when using clozapine.10,11 to 50 mg to achieve a daily dose of up to 300 mg within 5. -

(12) United States Patent (10) Patent No.: US 6,720,318 B2 Dib Et Al
USOO6720318B2 (12) United States Patent (10) Patent No.: US 6,720,318 B2 Dib et al. (45) Date of Patent: Apr. 13, 2004 (54) COMBINATION OF CYAMEMAZINE AND EP O5295OO 3/1993 AN ATYPCAL NEUROLEPTIC EP O586.191 9/1994 EP O733634 9/1996 (75) Inventors: Michel Dib, Paris (FR); Cyrille wo WO 52. AC Leperlier, Fontainebleau (FR) WO 96/30375 10/1996 (73) Assignee: Aventis Pharma S.A., Antony Cedex WO WO 98/43646is 10/1998go (FR) OTHER PUBLICATIONS (*) Notice: Subject to any disclaimer, the term of this patent is extended or adjusted under 35 Vanel et al., Eur. J. Clin. Pharmacol. (55, No. 9, 659–65, 1999) (abstract).* U.S.C. 154(b) by 0 days. Dietch, James T. et al., Aggressive Dyscontrol in Patients Treated with Benzodiazepines, J. Clin Psychiatry (1988, pp. (21) Appl. No.: 10/164,771 184-188, vol. 49:5). (22) Filed: Jun. 7, 2002 Overall, John E. et al., The Brief Psychiatric Rating Scale, Psychological Reports, (1962, pp. 799-812, vol. 10). (65) Prior Publication Data Senninger, J. L. et al., La Risperidone Dans Le Traitement de L'Agressivite Des Schizophrenes, Ann. Med. Psychol. US 2002/0183312 A1 Dec. 5, 2002 (1998, pp. 210-213, vol. 156 No. 3). Related U.S. Application Data Stimmel, Glen L., Benzodiazepines in Schizophrenia, Supplement to Pharmacotherapy (1996, pp. 148-151, vol. (63) Continuation of application No. PCT/FR00/03446, filed on 16). Dec. 8, 2000. Warneke, Lorne B., Benzodiazepines: Abuse and New Use, (30) Foreign Application Priority Data Can. J. Psychiatry (1991, pp. 194–205, vol. -

The Anticholinergic Impregnation Scale: Towards The
Therapie (2017) 72, 427—437 Available online at ScienceDirect www.sciencedirect.com CLINICAL PHARMACOLOGY The anticholinergic impregnation scale: Towards the elaboration of a scale adapted to prescriptions in French psychiatric settings L’échelle d’imprégnation anticholinergique : vers l’élaboration d’une échelle adaptée aux prescriptions en milieu psychiatrique franc¸ais a,b b,c,∗ Jeanne Briet , Hervé Javelot , c d Edwige Heitzmann , Luisa Weiner , c e Catherine Lameira , Philippe D’Athis , e a,b Marie Corneloup , Jean-Louis Vailleau a Pharmacy service, CHS de La Chartreuse, 21000 Dijon, France b PIC network (Psychiatrie Information Communication), EPSM Lille-Métropole, 59487 Armentières, France c Établissement public de santé Alsace Nord, 67170 Brumath, France d Psychiatry II and Inserm unit 1114, university hospital of Strasbourg, 67000 Strasbourg, France e Service of biostatistics and medical informatics, CHU de Dijon, 21000 Dijon, France Received 6 June 2016; accepted 23 December 2016 Available online 17 February 2017 KEYWORDS Summary Anticholinergics; Purpose. — Some drugs have anticholinergic activity and can cause peripheral or central side Anticholinergic drug effects. Several scales exist to evaluate the potential anticholinergic effect of prescribed drugs scale; but: (i) they are validated in the elderly and mainly assess the cognitive side effect of treat- Psychiatry ments; (ii) they do not concern some of the drugs frequently used in clinical psychiatry in France. The aim of our study is to develop a new scale, the anticholinergic impregnation scale (AIS), with drugs used in France and based on an assessment of the drugs used against peripheral anticholinergic adverse effects. ∗ Corresponding author. Clinical pharmacy service, Établissement public de santé Alsace Nord (EPS Alsace Nord), 141 avenue de Strasbourg, 67170 Brumath, France. -

Drug Interactions with Proton Pump Inhibitor Or Infectious Etiologies? Sophie Wagner, M
Confounding factors for variation of clozapine plasma levels: drug interactions with proton pump inhibitor or infectious etiologies? Sophie Wagner, M. G. Varet-Legros, Claudine Fabre, Jean Louis Montastruc, Haleh Bagheri To cite this version: Sophie Wagner, M. G. Varet-Legros, Claudine Fabre, Jean Louis Montastruc, Haleh Bagheri. Con- founding factors for variation of clozapine plasma levels: drug interactions with proton pump inhibitor or infectious etiologies?. European Journal of Clinical Pharmacology, Springer Verlag, 2010, 67 (5), pp.533-534. 10.1007/s00228-010-0925-z. hal-00638575 HAL Id: hal-00638575 https://hal.archives-ouvertes.fr/hal-00638575 Submitted on 6 Nov 2011 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Page 3 of 8 European Journal of Clinical Pharmacology 1 2 Confounding factors for variation of clozapine plasma levels: drug interactions with 3 proton pump inhibitor or infectious aetiologies? 4 S Wagner 1, MG Varet-Legros 2, C Fabre 3, JL Montastruc 1, H Bagheri 1. 5 6 1 Service de Pharmacologie Clinique, Centre midi-pyrénées -
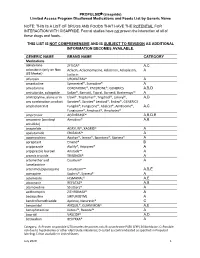
PROPULSID® (Cisapride) Limited Access Program
PROPULSID (cisapride) Limited Access Program Disallowed Medications and Foods List by Generic Name NOTE: THIS IS A LIST OF DRUGS AND FOODS THAT HAVE THE POTENTIAL FOR INTERACTION WITH CISAPRIDE. Formal studies have not proven the interaction of all of these drugs and foods. THIS LIST IS NOT COMPREHENSIVE AND IS SUBJECT TO REVISION AS ADDITIONAL INFORMATION BECOMES AVAILABLE. GENERIC NAME BRAND NAME CATEGORY Medications abiraterone ZYTIGA® A,C aclarubicin (only on Non Aclacin, Aclacinomycine, Aclacinon, Aclaplastin, A US Market) Jaclacin alfuzosin UROXATRAL® A amantadine Symmetrel®, Symadine® A amiodarone CORDARONE®, PACERONE®, GENERICS A,B,D amisulpride, sultopride Solian®, Barnotil, Topral, Barnetil, Barhemsys™ A amitriptyline, alone or in Elavil®, Tryptomer®, Tryptizol®, Laroxyl®, A,D any combination product Saroten®, Sarotex® Lentizol®, Endep®, GENERICS amphotericin B Fungilin®, Fungizone®, Abelcet®, AmBisome®, A,C Fungisome®, Amphocil®, Amphotec® amprenavir AGENERASE® A,B,D,E amsacrine (acridinyl Amsidine® A,B anisidide) anagrelide AGRYLIN®, XAGRID® A apalutamide ERLEADA™ A apomorphine Apokyn®, Ixense®, Spontane®, Uprima® A aprepitant Emend® B aripiprazole Abilify®, Aripiprex® A aripiprazole lauroxil Aristada™ A arsenic trioxide TRISENOX® A artemether and Coartem® A lumefantrine artenimol/piperaquine Eurartesim™ A,B,E asenapine Saphris®, Sycrest® A astemizole HISMANAL® A,E atazanavir REYATAZ® A,B atomoxetine Strattera® A azithromycin ZITHROMAX® A bedaquiline SIRTURO(TM) A bendroflumethiazide Aprinox, Naturetin® C benperidol ANQUIL®, -

Binding of Levomepromazine and Cyamemazine to Human Recombinant Dopamine Receptor Subtypes
Eur. J. Psychiat. Vol. 23, N.° 3, (147-152) 2009 Keywords: Clozapine; Cyamemazine; Dopamine receptor subtypes; Levomepromazine; Schizophre- nia. Binding of levomepromazine and cyamemazine to human recombinant dopamine receptor subtypes Lalit K. Srivastava*,** Neelakanta P.V. Nair*,** Samarthji Lal*,**,*** * Douglas Mental Health University Institute, Montreal, Quebec ** Department of Psychiatry, McGill University *** Department of Psychiatry, Montreal General Hospital, Montreal, Quebec CANADA ABSTRACT – Background and Objectives: Clozapine (CLOZ) and levomepromazine (LMP) improve treatment-resistant schizophrenia. The superior efficacy of CLOZ com- pared with other antipsychotic agents has been attributed to an effect on D1-like and D4 receptors. We examined the binding of LMP, CLOZ and cyamemazine (CMZ), a neu- roleptic analog of LMP, to human recombinant dopamine (rDA) receptor subtypes. Methods: Binding studies were performed on frozen membrane suspensions of human rDA receptor subtypes expressed in Sf9 cells. Results: (i) LMP has a high affinity (Ki, nM) for rD2 receptor subtypes (rD2L 8.6; rD2S 4.3; rD3 8.3; rD4.2 7.9); (ii) LMP and CLOZ have comparable affinities for the rD1 receptor (54.3 vs 34.6); (iii) CMZ has high affinities for rD2-like and rD1-like receptors (rD2L 4.6; rD2S 3.3; rD3 6.2; rD4.2 8.5; rD1 3.9; rD5 10.7); (iv) CMZ is 9 times more po- tent than CLOZ at the rD1 receptor and 5 times more potent than CLOZ at the rD4.2 re- ceptor; (v) CMZ has high affinities for rD1 and rD5 receptor subtypes compared with LMP and CLOZ. Conclusions: If D1 and D4 receptors are important sites for the unique action of CLOZ, the present study points to a need for clinical trials comparing CMZ with CLOZ in schizophrenia and in particular, treatment-resistant schizophrenia, especially given the risk for agranulocytosis with CLOZ.