Horizon Scanning Status Report June 2020 Prepared For: Patient-Centered Outcomes Research Institute 1828 L St., NW, Suite 900 Washington, DC 20036
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Optumrx Brand Pipeline Forecast
RxOutlook® 1st Quarter 2019 OptumRx brand pipeline forecast Route of Regulatory Estimated Specialty Orphan Drug name Generic name Company Drug class Therapeutic use administration status release date drug drug 2019 Possible launch date Ophthalmological DS-300 DS-300 Eton undisclosed SC Filed NDA 2019 unknown N disease anti-sclerostin Evenity romosozumab Amgen Osteoporosis SC Filed NDA 2/2019 Y N monoclonal antibody tetrahydrofolate iclaprim iclaprim Motif Bio Bacterial infections IV Filed NDA 2/13/2019 Y Y dehydrogenase inhibitor tazarotene/ IDP-118 Valeant retinoid/ corticosteroid Psoriasis TOP Filed NDA 2/15/2019 N N halobetasol adenosine deaminase Mavenclad cladribine Merck/ Teva resistant Multiple sclerosis PO Filed NDA 2/15/2019 Y N deoxyadenosine analog Lotemax Gel loteprednol Valeant corticosteroid Ocular inflammation OP Filed NDA 2/25/2019 N N Nex Gen etabonate turoctocog alfa glyco-PEGylated factor NN-7088 Novo Nordisk Hemophilia IV/SC Filed BLA 2/27/2019 Y N pegol VIII derivative selective sphingosine-1 BAF-312 siponimod Novartis phosphate receptor Multiple sclerosis PO Filed NDA 3/1/2019 Y N agonist midazolam midazolam UCB benzodiazepine Seizures Intranasal Filed NDA 3/1/2019 N Y (USL-261) XeriSol glucagon Xeris glucagon analog Diabetes mellitus SC Filed NDA 3/1/2019 N N Glucagon optum.com/optumrx 1 RxOutlook® 1st Quarter 2019 Route of Regulatory Estimated Specialty Orphan Drug name Generic name Company Drug class Therapeutic use administration status release date drug drug dopamine receptor JZP-507 sodium oxybate Jazz Narcolepsy -

Fecal Microbiota Transplantation in Inflammatory Bowel Disease: the Quest for the Holy Grail
REVIEW Fecal microbiota transplantation in inflammatory bowel disease: the quest for the holy grail B Pigneur1,2,3,* and H Sokol3,4,5,6,* Inflammatory bowel disease (IBD) is due to an aberrant immune response toward luminal antigens, probably commensal bacteria, in genetically susceptible subjects and is also influenced by environmental factors. An imbalanced intestinal microbiota known as ‘‘dysbiosis,’’ characterized by an increased proportion of pro-inflammatory microorganisms and a decreased proportion of anti-inflammatory microorganisms, has been repeatedly observed in IBD and is now recognized as a key factor in the gut inflammatory process. Fecal microbiota transplantation (FMT) has gained interest as a novel treatment option in IBD. The goal of FMT in IBD is not only to correct the dysbiosis, but also to restore a normal dialog between the host immune system and the microbiota. Data are still scarce, but the results of the first studies suggest that FMTcould be a promising therapy in IBD. More studies are needed to define the best indications, optimal timing, frequency, mode of delivery, and the optimal donor for each patient. INTRODUCTION has only recently been proven to be efficient in a randomized Although major progress has been achieved in recent years, the control trial.8 Following this study, FMT is now being evaluated pathogenesis of inflammatory bowel disease (IBD) has not yet in several other microbiota-driven diseases and particularly in been fully elucidated. It is generally accepted that IBD is caused IBD. CDI is a pure ecological problem characterized by a defect by an aberrant immune response toward luminal antigens, in gut microbiota barrier properties.9,10 IBD, however, is far most likely commensal bacteria, in genetically susceptible more complex and involves a deregulation of the host-microbes subjects and is also under the influence of environmental cross talk. -

Safety and Tolerability of Arimoclomol in Patients with Sporadic Inclusion Body Myositis: a Randomised, Double-Blind, Placebo-Controlled, Proof-Of-Concept Trial
Safety and tolerability of arimoclomol in patients with sporadic inclusion body myositis: a randomised, double-blind, placebo-controlled, proof-of-concept trial Sporadic inclusion body myositis (IBM) is the commonest idiopathic inflammatory myopathy (IIM) occurring in patients over the age of 50 years.1-4 The prevalence of IBM differs between different populations and ethnic groups, with estimates between 4.9-70.6 per million population.5-10 IBM is distinguished from the other IIM by early asymmetric finger flexor and knee extensor weakness (leading to loss of hand function and propensity to fall) and resistance to immunosuppressive therapy. Any skeletal muscle can be affected, including oesophageal and pharyngeal muscles. Late-stage disease can be characterized by very significant morbidity including disability and loss of quality of life. Death in IBM is related to malnutrition, cachexia, aspiration, respiratory infection and respiratory failure, as a consequence of dysphagia, severe global weakness and weakness of the respiratory muscles.1-4 In a Dutch cohort, end-of-life care interventions were used by 13% of patients with IBM, highlighting the morbidity experienced by these patients and the need of supportive and palliative care in this disease.2 The aetiopathogenesis of IBM remains uncertain. The varied pathological findings observed have driven a number of theories including viral infection, accumulation of toxic proteins, autoimmune attack, myonuclear degeneration, endoplasmic reticulum stress and impairment of autophagy and proteasomal proteolysis.11-15 Muscle biopsies from patients with IBM typically show several different pathological features broadly described as inflammatory or degenerative. Inflammatory features include endomysial infiltration by mononuclear cells (predominantly CD8+ T- cells), which surround and invade otherwise normal appearing muscle fibres, and overexpression of major histocompatibility complex (MHC) class I, which is not constitutively expressed by skeletal muscle. -

Parkinson Disease-Associated Cognitive Impairment
PRIMER Parkinson disease-associated cognitive impairment Dag Aarsland 1,2 ✉ , Lucia Batzu 3, Glenda M. Halliday 4, Gert J. Geurtsen 5, Clive Ballard 6, K. Ray Chaudhuri 3 and Daniel Weintraub7,8 Abstract | Parkinson disease (PD) is the second most common neurodegenerative disorder, affecting >1% of the population ≥65 years of age and with a prevalence set to double by 2030. In addition to the defining motor symptoms of PD, multiple non-motor symptoms occur; among them, cognitive impairment is common and can potentially occur at any disease stage. Cognitive decline is usually slow and insidious, but rapid in some cases. Recently, the focus has been on the early cognitive changes, where executive and visuospatial impairments are typical and can be accompanied by memory impairment, increasing the risk for early progression to dementia. Other risk factors for early progression to dementia include visual hallucinations, older age and biomarker changes such as cortical atrophy, as well as Alzheimer-type changes on functional imaging and in cerebrospinal fluid, and slowing and frequency variation on EEG. However, the mechanisms underlying cognitive decline in PD remain largely unclear. Cortical involvement of Lewy body and Alzheimer-type pathologies are key features, but multiple mechanisms are likely involved. Cholinesterase inhibition is the only high-level evidence-based treatment available, but other pharmacological and non-pharmacological strategies are being tested. Challenges include the identification of disease-modifying therapies as well as finding biomarkers to better predict cognitive decline and identify patients at high risk for early and rapid cognitive impairment. Parkinson disease (PD) is the most common movement The full spectrum of cognitive impairment occurs in disorder and the second most common neurodegenera individuals with PD, from subjective cognitive decline tive disorder after Alzheimer disease (AD). -

Therapeutic Class Overview Colony Stimulating Factors
Therapeutic Class Overview Colony Stimulating Factors Therapeutic Class Overview/Summary: This review will focus on the granulocyte colony stimulating factors (G-CSFs) and granulocyte- macrophage colony stimulating factors (GM-CSFs).1-5 Colony-stimulating factors (CSFs) fall under the naturally occurring glycoprotein cytokines, one of the main groups of immunomodulators.6 In general, these proteins are vital to the proliferation and differentiation of hematopoietic progenitor cells.6-8 The G- CSFs commercially available in the United States include pegfilgrastim (Neulasta®), filgrastim (Neupogen®), filgrastim-sndz (Zarxio®), and tbo-filgrastim (Granix®). While filgrastim-sndz and tbo- filgrastim are the same recombinant human G-CSF as filgrastim, only filgrastim-sndz is considered a biosimilar drug as it was approved through the biosimilar pathway. At the time tbo-filgrastim was approved, a regulatory pathway for biosimilar drugs had not yet been established in the United States and tbo-filgrastim was filed under its own Biologic License Application.9 Only one GM-CSF is currently available, sargramostim (Leukine). These agents are Food and Drug Administration (FDA)-approved for a variety of conditions relating to neutropenia or for the collection of hematopoietic progenitor cells for collection by leukapheresis.1-5 Due to the pathway taken, tbo-filgrastim does not share all of the same indications as filgrastim and these two products are not interchangeable. It is important to note that although filgrastim-sndz is a biosimilar product, and it was approved with all the same indications as filgrastim at the time, filgrastim has since received FDA-approval for an additional indication that filgrastim-sndz does not have, to increase survival in patients with acute exposure to myelosuppressive doses of radiation.1-3A complete list of indications for each agent can be found in Table 1. -

2017 ANNUAL REPORT | Translating SCIENCE • Transforming LIVES OUR COMMITMENT Make Every Day Count at PTC, Patients Are at the Center of Everything We Do
20 YEARS OF COMMITMENT 2017 ANNUAL REPORT | Translating SCIENCE • Transforming LIVES OUR COMMITMENT Make every day count At PTC, patients are at the center of everything we do. We have the opportunity to support patients and families living with rare disorders through their journey. We know that every day matters and we are committed to making a difference. OUR SCIENCE Our scientists are finding new ways to regulate biology to control disease We have several scientific research platforms focused on modulating protein expression within the cell that we believe have the potential to address many rare genetic disorders. OUR PEOPLE Care for each other, our community, and for the needs of our patients At PTC, we are looking at drug discovery and development in a whole new light, bringing new technologies and approaches to developing medicines for patients living with rare disorders and cancer. We strive every day to be better than we were the day before. At PTC Therapeutics, it is our mission to provide access to best-in-class treatments for patients who have an unmet need. We are a science-led, global biopharmaceutical company focused on the discovery, development and commercialization of clinically-differentiated medicines that provide benefits to patients with rare disorders. Founded 20 years ago, PTC Therapeutics has successfully launched two rare disorder products and has a global commercial footprint. This success is the foundation that drives investment in a robust pipeline of transformative medicines and our mission to provide access to best-in-class treatments for patients who have an unmet medical need. As we celebrate our 20th year of bringing innovative therapies to patients affected by rare disorders, we reflect on our unwavering commitment to our patients, our science and our employees. -
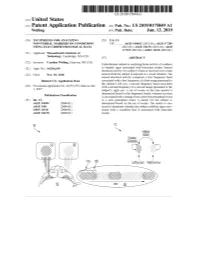
ANNNNNNNNNNNNNNNNNNNN 100A 006 Left Eye Input Right Eye Input
US 20190175049A1 ( 19) United States (12 ) Patent Application Publication (10 ) Pub. No. : US 2019 /0175049 A1 Welling ( 43 ) Pub . Date : Jun . 13 , 2019 ( 54 ) TECHNIQUES FOR ANALYZING (52 ) U . S . CI. NON -VERBAL MARKERS OF CONDITIONS CPC . .. A61B 5 /04842 (2013 . 01 ) ; A61B 5 / 7289 USING ELECTROPHYSIOLOGICAL DATA (2013 . 01) ; A61B 5 /0478 ( 2013 .01 ) ; A61B 5 /7225 ( 2013. 01 ) ; G06N 20 / 10 (2019 .01 ) (71 ) Applicant: Massachusetts Institute of Technology , Cambridge , MA (US ) ( 57 ) ABSTRACT (72 ) Inventor : Caroline Welling, Hanover, NH (US ) Embodiments related to analyzing brain activity of a subject to identify signs associated with binocular rivalry . Sensed ( 21 ) Appl. No. : 16 / 206, 639 electrical activity of a subject' s brain is received over a time period while the subject is exposed to a visual stimulus. The ( 22 ) Filed : Nov. 30 , 2018 sensed electrical activity comprises a first frequency band Related U . S . Application Data associated with a first frequency of a first image presented to the subject ' s left eye , a second frequency band associated (60 ) Provisional application No .62 / 593 , 535, filed on Dec . with a second frequency of a second image presented to the 1 , 2017 subject ' s right eye . A set of events in the time period is determined based on the frequency bands, wherein an event Publication Classification is associated with a change from a previous perceptual event (51 ) Int. Ci. to a new perceptual event. A metric for the subject is A61B 5 /0484 ( 2006 .01 ) determined based on the set of events . The metric is ana A61B 5 /00 ( 2006 .01 ) lyzed to determine whether the subject exhibits signs asso GO6N 20 / 10 (2006 .01 ) ciated with a condition that is associated with binocular A61B 5 /0478 ( 2006 .01 ) rivalry . -

Relmada Therapeutics Announces Notice of Acceptance of Key Patent in Europe Covering NMDA Receptor Antagonist D- Methadone for Treatment of Psychiatric Symptoms
January 9, 2018 Relmada Therapeutics Announces Notice of Acceptance of Key Patent in Europe Covering NMDA Receptor Antagonist d- Methadone for Treatment of Psychiatric Symptoms Patent significantly expands Relmada intellectual property protection and positions company to target global commercial opportunities for wide range of psychiatric disorders. NEW YORK, Jan. 9, 2018 /PRNewswire/ -- Relmada Therapeutics, Inc. (OTCQB: RLMD), a clinical-stage company developing novel therapies for the treatment of central nervous system (CNS) diseases, announced today that the European Patent Office has issued a notice of allowance for patent application number 13773543.7 for "D-methadone for the treatment of psychiatric symptoms." The patent provides broad coverage in Europe for d- Methadone (dextromethadone, REL-1017), a novel N-methyl-D-aspartate (NMDA) receptor antagonist, for the treatment of symptoms associated with a range of psychological and psychiatric disorders including depression, anxiety, fatigue and mood instability. "The allowance of this key patent significantly strengthens our IP position and the global commercial opportunities in our d-Methadone program," said Sergio Traversa, CEO of Relmada Therapeutics. "We look forward to advancing our current development programs and to working to identify potential new areas of unmet need where d-Methadone can deliver benefits to patients in the years ahead." The NMDA receptor is a predominant molecular device for controlling synaptic plasticity and memory function and affects the transfer of electrical signals between neurons in the brain and in the spinal column. Based on their mechanism of action, a range of NMDA receptor antagonists (chemicals that deactivate the NMDA receptor) such as d-Methadone are under consideration as potential therapeutic agents for the treatment of many CNS conditions including some psychiatric disorders. -

Relmada Announces FDA Fast Track Designation for D-Methadone for Adjunctive Treatment of Major Depressive Disorder
April 13, 2017 Relmada Announces FDA Fast Track Designation for d-Methadone for Adjunctive Treatment of Major Depressive Disorder NEW YORK, April 13, 2017 /PRNewswire/ -- Relmada Therapeutics, Inc. (OTCQB: RLMD), a clinical-stage company developing novel therapies for the treatment of central nervous system (CNS) diseases, today announced that the U.S. Food and Drug Administration (FDA) has granted Fast Track designation for d-Methadone (REL-1017 dextromethadone), the company's novel N-methyl-D-aspartate (NMDA) receptor antagonist in development for the adjunctive treatment of major depressive disorder. Fast Track designation is a process designed to facilitate the development and expedite the review of drugs to treat serious conditions and fill an unmet medical need. The purpose, according to the FDA, is to get important new drugs to the patient earlier. Drugs that receive Fast Track designation may be eligible for more frequent meetings and written communications with the FDA, accelerated review and priority approval, and rolling New Drug Application review. "Treatment of depression continues to be a significant challenge in healthcare affecting millions of patients around the world," said Richard Mangano, Ph.D., chief scientific officer of Relmada. "The designation of Fast Track status by the FDA is further validation of the potential for d-Methadone to represent a major advance in treatment that can help patients with inadequate response to the current standard of care. We look forward to working with the FDA to advance the development program for d-Methadone and an expedited regulatory process." Relmada is planning to advance the development program for REL-1017 to a phase 2a randomized, double-blind, placebo-controlled study in patients with major depressive disorder. -

Targeted Therapies in Advanced Cholangiocarcinoma: a Focus on FGFR Inhibitors
medicina Review Targeted Therapies in Advanced Cholangiocarcinoma: A Focus on FGFR Inhibitors Alessandro Rizzo Department of Experimental, Diagnostic and Specialty Medicine, S. Orsola-Malpighi University Hospital, 40138 Bologna, Italy; [email protected] Abstract: Despite advanced diseases continuing to be associated with grim prognoses, the past decade has witnessed the advent of several novel treatment options for cholangiocarcinoma (CCA) patients. In fact, CCA has emerged as a heterogeneous group of malignancies harboring potentially druggable mutations in approximately 50% of cases, and thus, molecularly targeted therapies have been actively explored in this setting. Among these, fibroblast growth factor receptor (FGFR) inhibitors have reported important results, as witnessed by the FDA approval of pemigatinib in previously treated metastatic CCA patients harboring FGFR2 fusion or other rearrangements. Herein, we provide an overview of available evidence on FGFR inhibitors in CCA, especially focusing on the development, pitfalls and challenges of emerging treatments in this setting. Keywords: FGFR; cholangiocarcinoma; targeted therapies; intrahepatic cholangiocarcinoma; pemi- gatinib 1. Introduction Citation: Rizzo, A. Targeted Cholangiocarcinoma (CCA) encompasses a group of heterogeneous, rare and aggres- Therapies in Advanced sive malignancies, including intrahepatic cholangiocarcinoma (iCCA) and extrahepatic Cholangiocarcinoma: A Focus on cholangiocarcinoma (eCCA), with the latter further subclassified into perihilar (pCCA) FGFR Inhibitors. Medicina 2021, 57, and distal (dCCA) cholangiocarcinoma [1–3]. CCAs account for approximately 3% of 458. https://doi.org/10.3390/ all gastrointestinal cancers worldwide and 10–15% of all primary liver tumors [4–6]. As medicina57050458 suggested by several studies, these subgroups of hepatobiliary tumors not only develop from different anatomical locations, but vary widely in terms of epidemiology, biology, Academic Editor: Zygmunt Warzecha prognosis, and etiology [7–9]. -

Effect of Repeated Gaboxadol Administration on Night Sleep and Next-Day Performance in Healthy Elderly Subjects
Neuropsychopharmacology (2005) 30, 833–841 & 2005 Nature Publishing Group All rights reserved 0893-133X/05 $30.00 www.neuropsychopharmacology.org Effect of Repeated Gaboxadol Administration on Night Sleep and Next-Day Performance in Healthy Elderly Subjects 1 2 ,3 1 Stefan Mathias , Josef Zihl , Axel Steiger* and Marike Lancel 1Section of Sleep Pharmacology, Max-Planck-Institute of Psychiatry, Munich, Germany; 2Section of Neuropsychology, Max-Planck-Institute of Psychiatry, Munich, Germany; 3Department of Psychiatry, Max-Planck-Institute of Psychiatry, Munich, Germany Aging is associated with dramatic reductions in sleep continuity and sleep intensity. Since gaboxadol, a selective GABAA receptor agonist, has been demonstrated to improve sleep consolidation and promote deep sleep, it may be an effective hypnotic, particularly for elderly patients with insomnia. In the present study, we investigated the effects of subchronic gaboxadol administration on nocturnal sleep and its residual effects during the next days in elderly subjects. This was a randomized, double-blind, placebo-controlled, balanced crossover study in 10 healthy elderly subjects without sleep complaints. The subjects were administered either placebo or 15 mg gaboxadol hydrochloride at bedtime on three consecutive nights. Sleep was recorded during each night from 2300 to 0700 h and tests assessing attention (target detection, stroop test) and memory function (visual form recognition, immediate word recall, digit span) were applied at 0900, 1400, and 1700 h during the following days. Compared with placebo, gaboxadol significantly shortened subjective sleep onset latency and increased self-rated sleep intensity and quality. Polysomnographic recordings showed that it significantly decreased the number of awakenings, the amount of intermittent wakefulness, and stage 1, and increased slow wave sleep and stage 2. -

Duchenne Muscular Dystrophy (DMD) Agents
Therapeutic Class Overview Duchenne muscular dystrophy (DMD) Agents INTRODUCTION • Duchenne muscular dystrophy (DMD) is 1 of 4 conditions known as dystrophinopathies, which are inherited, X-linked myopathic disorders due to a defect in the dystrophin gene that results in the primary pathologic process of muscle fiber degradation. The hallmark symptom is progressive weakness (Darras 2018[a], Darras 2018[b], Muscular Dystrophy Association [MDA] 2019). The other 3 conditions include: Becker muscular dystrophy (BMD), which is a mild form of DMD; an intermediate presentation between BMD and DMD; and DMD-associated dilated cardiomyopathy, which has little or no clinical ○ skeletal or muscle disease (MDA 2019). • DMD symptom onset is in early childhood, usually between the ages of 2 and 3 years old. The proximal muscles are affected first, followed by the distal limb muscles. Generally, the lower external muscles will be affected before the upper. The affected child may have difficulties jumping, walking, and running (MDA 2019). • The prevalence of DMD ranges from 1 to 2 per 10,000 live male births; female-manifesting carriers are rarer, but can present with a range of symptoms that vary in their severities (Birnkrant et al 2018, Darras 2018[a], Emflaza Food and Drug Administration [FDA] Medical Review 2017). • The clinical course and lifespan of patients with DMD is relatively short. Individuals are usually confined to a wheelchair by age 13, and many die in their late teens or twenties from respiratory insufficiency or cardiomyopathy. Although survival until adulthood is more common now, very few patients survive past the 3rd decade (Darras 2018[a]).