Molecular Ecology
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Strategies for Complete Plastid Genome Sequencing
Edinburgh Research Explorer Strategies for complete plastid genome sequencing Citation for published version: Twyford, A & Ness, R 2017, 'Strategies for complete plastid genome sequencing', Molecular Ecology Resources, vol. 17, no. 5, pp. 858-868. https://doi.org/10.1111/1755-0998.12626 Digital Object Identifier (DOI): 10.1111/1755-0998.12626 Link: Link to publication record in Edinburgh Research Explorer Document Version: Publisher's PDF, also known as Version of record Published In: Molecular Ecology Resources Publisher Rights Statement: © 2016 The Authors. Molecular Ecology Resources Published by John Wiley & Sons Ltd. This is an open access article under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in any medium, provided the original work is properly cited. General rights Copyright for the publications made accessible via the Edinburgh Research Explorer is retained by the author(s) and / or other copyright owners and it is a condition of accessing these publications that users recognise and abide by the legal requirements associated with these rights. Take down policy The University of Edinburgh has made every reasonable effort to ensure that Edinburgh Research Explorer content complies with UK legislation. If you believe that the public display of this file breaches copyright please contact [email protected] providing details, and we will remove access to the work immediately and investigate your claim. Download date: 28. Sep. 2021 Molecular Ecology Resources (2016) doi: 10.1111/1755-0998.12626 Strategies for complete plastid genome sequencing ALEX D. TWYFORD* and ROB W. NESS† *Institute of Evolutionary Biology, Ashworth Laboratories, University of Edinburgh, Edinburgh EH9 3FL, UK, †Department of Biology, University of Toronto Mississauga, Mississauga, ON, Canada Abstract Plastid sequencing is an essential tool in the study of plant evolution. -

Christopher William Dick University of Michigan Department of Ecology and Evolutionary Biology Biological Science Building Room 2068, 1105 N
Christopher William Dick University of Michigan Department of Ecology and Evolutionary Biology Biological Science Building Room 2068, 1105 N. University Ave Ann Arbor, MI 48109-1085 734-764-9408 (voice) 734-763-0544 (fax) [email protected] orcid.org/0000-0001-8745-9137 http://sites.lsa.umich.edu/cwdick-lab/ Education 1999 Ph.D. Department of Organismic and Evolutionary Biology, Harvard University 1997 M.A. Department of Organismic and Evolutionary Biology, Harvard University 1990 B.A. Hampshire College, Amherst, Massachusetts Present Appointments 2017- Associate Chair for Museum Collections (UM Herbarium and Museum of Zoology) 2016- Professor and Curator, EEB Department, University of Michigan 2006- Research Associate, Smithsonian Tropical Research Institute Previous Appointments 2015-2017 Associate Chair for Museum Collections (UM Herbarium) 2014-2017 Director of the Edwin S. George Reserve, University of Michigan 2011-2016 Associate Professor and Associate Curator, University of Michigan 2012-2013 Acting Director of the U-M Herbarium/ Associate Chair for Museum Collections 2005-2011 Assistant Professor and Assistant Curator, University of Michigan 2002-2005 Tupper Postdoctoral Fellow, Smithsonian Tropical Research Institute 2001-2002 Mellon Postdoctoral Fellow, Smithsonian Tropical Research Institute 1999-2001 Molecular Evolution Postdoctoral Fellow, Smithsonian Tropical Research Institute 1992-1999 Graduate student, Harvard University 1992 Botanical Intern, Biological Dynamics of Forest Fragments Project, Manaus, Brazil 1991 Field Biologist, U.S. Forest Service, Globe Forest District, Arizona Publication List (*Student or post-doc working in the Dick lab) for citation history see https://scholar.google.com/citations?user=7iy44V4AAAAJ 1. Mander, L., C. Parins-Fukuchi*, C. W. Dick, S. W. Punyasena, C. Jaramillo (in review) Phylogenetic and ecological drivers of pollen morphological diversity in a Neotropical rainforest. -

Population Size and Genetic Diversity of Nigerian Lions (Panthera Leo)
POPULATION SIZE AND GENETIC DIVERSITY OF NIGERIAN LIONS (PANTHERA LEO) POPULATION SIZE AND GENETIC DIVERSITY OF NIGERIAN LIONS (PANTHERA LEO) Talatu Tende AKADEMISK AVHANDLING Som för av filosofie doktorsexamen vid naturvetenskapliga fakulteten, Lunds universitet, kommer att offentligen försvaras i Blå Hallen, Ekologihuset, Lund, Fredagen den 31 January 2014, kl.9:00. ACADEMIC DISSERTATION Presented in fulfilment of the requirements for the degree of Philosophie Doctor at the Faculty of Science, Lund University, to be defended publicly in the Blue Hall, Ecology Building, Sölvegatan 37, Lund, Sweden, Friday 31st January 2014, 9 AM. Fakultetsopponent: Göran Spong, Swedish University of Agricultural Sciences, Umeå POPULATION SIZE AND GENETIC DIVERSITY OF NIGERIAN LIONS (PANTHERA LEO) Talatu Tende A doctoral thesis at a university in Sweden is produced either as a monograph or as a collection of papers. In the latter case, the introductory part constitutes the formal thesis, which summa- rizes the accompanying papers. These have either already been published or are manuscripts at various stages (in press, submitted or in ms). Copyright © Talatu Tende Department of Biology | Lund University Cover art and intro chapter title art: Stina Andersson Layout and formatting: Katarina Eriksson Printed in Sweden at Media Tryck, Lund, 2013 ISBN: 978-91-7473-773-8 CONTENTS List of papers 8 List of contribution 8 Introduction 11 References 30 Popular Summary 37 Acknowledgments 39 Papers Paper I 45 Paper II 57 Paper III 73 Paper IV 85 Paper V 91 7 LIST OF PAPERS This thesis is based on the following papers which are referred to by their Roman numerals. I Ulf Ottosson, Talatu Tende, Christian Hjort & Bengt Hansson. -

What Is a Population? an Empirical Evaluation of Some Genetic Methods for Identifying the Number of Gene Pools and Their Degree of Connectivity
University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln Publications, Agencies and Staff of the U.S. Department of Commerce U.S. Department of Commerce 2006 What is a population? An empirical evaluation of some genetic methods for identifying the number of gene pools and their degree of connectivity Robin Waples NOAA, [email protected] Oscar Gaggiotti Université Joseph Fourier, [email protected] Follow this and additional works at: https://digitalcommons.unl.edu/usdeptcommercepub Waples, Robin and Gaggiotti, Oscar, "What is a population? An empirical evaluation of some genetic methods for identifying the number of gene pools and their degree of connectivity" (2006). Publications, Agencies and Staff of the U.S. Department of Commerce. 463. https://digitalcommons.unl.edu/usdeptcommercepub/463 This Article is brought to you for free and open access by the U.S. Department of Commerce at DigitalCommons@University of Nebraska - Lincoln. It has been accepted for inclusion in Publications, Agencies and Staff of the U.S. Department of Commerce by an authorized administrator of DigitalCommons@University of Nebraska - Lincoln. Molecular Ecology (2006) 15, 1419–1439 doi: 10.1111/j.1365-294X.2006.02890.x INVITEDBlackwell Publishing Ltd REVIEW What is a population? An empirical evaluation of some genetic methods for identifying the number of gene pools and their degree of connectivity ROBIN S. WAPLES* and OSCAR GAGGIOTTI† *Northwest Fisheries Science Center, 2725 Montlake Blvd East, Seattle, WA 98112 USA, †Laboratoire d’Ecologie Alpine (LECA), Génomique des Populations et Biodiversité, Université Joseph Fourier, Grenoble, France Abstract We review commonly used population definitions under both the ecological paradigm (which emphasizes demographic cohesion) and the evolutionary paradigm (which emphasizes reproductive cohesion) and find that none are truly operational. -

Information Theory Broadens the Spectrum of Molecular Ecology and Evolution
Please do not remove this page Information Theory Broadens the Spectrum of Molecular Ecology and Evolution Sherwin, William B.; Chao, Anne; Jost, Lou; et.al. https://scholarship.libraries.rutgers.edu/discovery/delivery/01RUT_INST:ResearchRepository/12643432920004646?l#13643526500004646 Sherwin, W. B., Chao, A., Jost, L., & Smouse, P. E. (2017). Information Theory Broadens the Spectrum of Molecular Ecology and Evolution. In Trends in Ecology And Evolution (Vol. 32, Issue 12, pp. 948–963). Rutgers University. https://doi.org/10.7282/T3Z89GD4 This work is protected by copyright. You are free to use this resource, with proper attribution, for research and educational purposes. Other uses, such as reproduction or publication, may require the permission of the copyright holder. Downloaded On 2021/09/24 16:12:15 -0400 1 Information Theory Broadens the Spectrum of 2 Molecular Ecology and Evolution 3 4 5 W.B. Sherwin 1 * 6 A. Chao 2 7 L. Jost 3 8 P.E. Smouse 4 9 10 Addresses: 11 1 Evolution and Ecology Research Centre, School of Biological Earth and Environmental Science, 12 University of New South Wales, Sydney NSW 2052 AUSTRALIA. 13 and 14 Murdoch University Cetacean Research Unit, Murdoch University, South Road, Murdoch, WA 6150 15 AUSTRALIA. 16 2 Institute of Statistics, National Tsing Hua University, Hsin-Chu 30043, Taiwan. 17 3 EcoMinga Foundation, Via a Runtun, Baños, Tungurahua, Ecuador 18 4 Department of Ecology, Evolution and Natural Resources, School of Environmental and Biological 19 Sciences, Rutgers University, New Brunswick, NJ 08901-8551, -

Molecular Ecology (2006) 15, 1561–1576 Doi: 10.1111/J.1365-294X.2006.02877.X
Molecular Ecology (2006) 15, 1561–1576 doi: 10.1111/j.1365-294X.2006.02877.x GeneticBlackwell Publishing Ltd diversity, population structure, effective population size and demographic history of the Finnish wolf population J. ASPI,* E. ROININEN,* M. RUOKONEN,* I. KOJOLA† and C. VILÀ‡ *Department of Biology, University of Oulu, P. O. Box 3000, FIN-90014, Oulu, Finland, †Finnish Game and Fisheries Research Institute, Oulu Game and Fisheries Research, Tutkijantie 2 E, FIN-90570 Oulu, Finland, ‡Department of Evolutionary Biology, Uppsala University, Norbyvägen 18D, S-752 36 Uppsala, Sweden Abstract The Finnish wolf population (Canis lupus) was sampled during three different periods (1996–1998, 1999–2001 and 2002–2004), and 118 individuals were genotyped with 10 micro- satellite markers. Large genetic variation was found in the population despite a recent demographic bottleneck. No spatial population subdivision was found even though a significant negative relationship between genetic relatedness and geographic distance suggested isolation by distance. Very few individuals did not belong to the local wolf population as determined by assignment analyses, suggesting a low level of immigration in the population. We used the temporal approach and several statistical methods to estimate the variance effective size of the population. All methods gave similar estimates of effective population size, approximately 40 wolves. These estimates were slightly larger than the estimated census size of breeding individuals. A Bayesian model based on Markov chain Monte Carlo simulations indicated strong evidence for a long-term population decline. These results suggest that the contemporary wolf population size is roughly 8% of its historical size, and that the population decline dates back to late 19th century or early 20th century. -

Divergence, Gene Flow, and Speciation in Eight Lineages of Trans‐Beringian Birds
Received: 2 May 2019 | Revised: 22 July 2020 | Accepted: 27 July 2020 DOI: 10.1111/mec.15574 ORIGINAL ARTICLE Divergence, gene flow, and speciation in eight lineages of trans-Beringian birds Jessica F. McLaughlin1,2 | Brant C. Faircloth3 | Travis C. Glenn4 | Kevin Winker1 1University of Alaska Museum, Fairbanks, AK, USA Abstract 2Sam Noble Oklahoma Museum of Natural Determining how genetic diversity is structured between populations that span the History, Norman, OK, USA divergence continuum from populations to biological species is key to understanding 3Department of Biological Sciences and Museum of Natural Science, Louisiana State the generation and maintenance of biodiversity. We investigated genetic divergence University, Baton Rouge, LA, USA and gene flow in eight lineages of birds with a trans-Beringian distribution, where 4 Department of Environmental Health Asian and North American populations have likely been split and reunited through Science and Institute of Bioinformatics, University of Georgia, Athens, GA, USA multiple Pleistocene glacial cycles. Our study transects the speciation process, in- cluding eight pairwise comparisons in three orders (ducks, shorebirds and passer- Correspondence Kevin Winker, University of Alaska Museum, ines) at population, subspecies and species levels. Using ultraconserved elements 907 Yukon Drive, Fairbanks, AK 99775, USA. (UCEs), we found that these lineages represent conditions from slightly differentiated Email: [email protected] populations to full biological species. Although allopatric speciation is considered Funding information the predominant mode of divergence in birds, all of our best divergence models in- National Science Foundation, Grant/Award Number: DEB-1242267-1242241-1242260 cluded gene flow, supporting speciation with gene flow as the predominant mode in Beringia. -

A Brief Guide to Conservation Biology Resources
A Brief Guide to Conservation Biology Resources. Compiled by Mark Schwartz, Elizabeth van Mantgem and Sara Kalmanovitz Department of Environmental Science & Policy. University of California, Davis CA 95616 January, 2000 for Joel Pagel of Region 5, USDA Forest Service. There has been a veritable explosion of resources over the past 10 years for conservation biologists. What follows is not meant to be a comprehensive list. Instead, this constitutes what we think are highlights in a very large literature. In particular, the journal articles cited are a combination of classic or benchmark papers in the field and a somewhat more eclectic suite of recent papers that represent examples of the application of various conservation strategies. 1. Journals General Conservation Journals: Biodiversity and Conservation. Bi-monthly. Chapman & Hall, London. http://www.wkap.nl/jrnltoc.htm/0960-3115 Biological Conservation. Monthly. Elsevier, Oxford, England.. Conservation Biology. Quarterly. Blackwell Scientific Publications, Cambridge, MA. Journal of the Society for Conservation Biology. Conservation Ecology. Electronic journal of the Ecological Society of America. http://www.consecol.org/Journal/ Ecological Applications. Quarterly. Ecological Society of America. http://www.jstor.org/journals/10510761.html Journal of Applied Ecology. Blackwell Scientific Publications Oxford [Eng.]. Journal of the British Ecological Society Journal of Biogeography. Bimonthly. Blackwell Scientific Publications, Oxford, Eng. Restoration Ecology. Quarterly Blackwell Scientific Publications. Cambridge, MA. Journal of the Society for Ecological Restoration. In addition, many taxon specific journals publish lots of conservation. For example: Aquatic Conservation. Quarterly, Wiley, New York. http://www.interscience.wiley.com/jpages/1052-7613/ Aquatic Ecology. Quarterly. Kluwer Academic Publishers, Dordrecht. http://www.wkap.nl/jrnltoc.htm/1386-2588 Audobon Wildlife Report. -

Ecological Divergence and Speciation in Common Bottlenose Dolphins in the Western South Atlantic
Received: 15 April 2019 | Revised: 1 November 2019 | Accepted: 28 November 2019 DOI: 10.1111/jeb.13575 SPECIAL ISSUE Ecological divergence and speciation in common bottlenose dolphins in the western South Atlantic Ana P. B. Costa1 | Pedro F. Fruet2,3,4,5 | Eduardo R. Secchi3 | Fábio G. Daura-Jorge6 | Paulo C. Simões-Lopes6 | Juliana C. Di Tullio2,3,4 | Patricia E. Rosel7 1Department of Biology, University of Louisiana at Lafayette, Lafayette, LA, USA Abstract 2Museu Oceanográfico ‘Prof. Eliézer C. Rios’, Coastal and offshore ecotypes of common bottlenose dolphins have been recognized Universidade Federal do Rio Grande, Rio in the western South Atlantic, and it is possible that trophic niche divergence associ- Grande, Brazil 3Laboratório de Ecologia e Conservação da ated with social interactions is leading them to genetic and phenotypic differentia- Megafauna Marinha (EcoMega), Instituto de tion. The significant morphological differentiation observed between these ecotypes Oceanografia, Universidade Federal do Rio Grande, Rio Grande, Brazil suggests they represent two different subspecies. However, there is still a need to 4Kaosa, Rio Grande, Brazil investigate whether there is congruence between morphological and genetic data 5Centro Nacional de Pesquisa e to rule out the possibility of ecophenotypic variation accompanied by gene flow. Conservação de Mamíferos Aquáticos – Mitochondrial DNA (mtDNA) control region sequence data and 10 microsatellite loci ICMBio/CMA, Santos, Brazil 6Laboratório de Mamíferos Aquáticos collected from stranded and biopsied dolphins sampled in coastal and offshore wa- (LAMAQ), Departamento de Ecologia e ters of Brazil as well as 106 skulls for morphological analyses were used to determine Zoologia, Universidade Federal de Santa Catarina, Florianópolis, Brazil whether the morphological differentiation was supported by genetic differentiation. -

Molecular Ecology - BIOLOGY 473 Fall 2008 - Drs
Molecular Ecology - BIOLOGY 473 Fall 2008 - Drs. Sergio Escorza Trevino & Elizabeth Torres LECTURE: MW 11:40 - 1:20 PM = BIOS-244 = CALL # 14788-01 COURSE DESCRIPTION: Applications of molecular tools to the study of ecology and evolution. Includes techniques, analytical methods, and case studies in conservation genetics, population genetics, and ecology. LECTURE SCHEDULE: Date Lecture Reading Assignment Mon. Sept. 22 Introduction Ch 1 Wed. Sept. 24 Molecular Markers 1 Ch 2 Mon. Sept. 29 Molecular Markers II P1 Ch 2 Wed. Oct. 1 Genetic Diversity Q1 Ch 3 Mon. Oct. 6 Population Genetics I Q2 Ch 4 Wed. Oct. 8 Population Genetics II P2 Ch 4 Mon. Oct. 13 MIDTERM I Wed. Oct. 15 Molecular Systematics I Q3 Journal Article Mon. Oct. 20 Molecular Systematics II P3 Journal Article Wed. Oct. 22 Phylogeography I Q4 Ch 5 Mon. Oct. 27 Phylogeography II Ch 5 Wed. Oct. 29 Species Identification Journal Article Mon. Nov. 3 Behavioral Ecology P4 Ch 6 Wed. Nov. 5 Conservation Genetics I Q5 Ch 7 Mon. Nov 10 MIDTERM II Wed. Nov. 12 Conservation Genetics II P5 Ch 7 Mon. Nov. 17 Conservation Genetics III Ch 7 Wed. Nov. 19 Ancient DNA Studies Journal Article Mon. Nov. 24 Applications I Ch 8 Wed. Nov. 26 Applications II Ch 8 Mon. Dec. 1 FINAL EXAM 10:45 am -1:15 pm TEXTBOOK: Molecular Ecology, 1st Edition, Joanna Freeland, 2006 Fall Quarter, 2008 Schedule of Classes - University Policies on Cheating and Plagiarism COURSE PREREQUISITE: Completion of Biology 360 (General Ecology) or equivalent. OFFICE HOURS AND CONTACT INFO: Dr. Elizabeth Torres: M 2-3pm; W 1:30-2:30pm La Kretz Hall 313; Biology Office: (323) 343-2050 Fax: (323) 343-6451; e-mail: [email protected] Please do not leave phone messages for Dr. -

Molecular Ecology Resources (2014) Doi: 10.1111/1755-0998.12301
Molecular Ecology Resources (2014) doi: 10.1111/1755-0998.12301 Transcriptome resources for the white-footed mouse (Peromyscus leucopus): new genomic tools for investigating ecologically divergent urban and rural populations STEPHEN E. HARRIS,* RACHEL J. O’NEILL† and JASON MUNSHI-SOUTH‡ *Program in Ecology, Evolutionary Biology, & Behavior, The Graduate Center, City University of New York (CUNY), New York, NY 10016, USA, †Molecular & Cell Biology, University of Connecticut, Storrs, CT 06269, USA, ‡Louis Calder Center-Biological Field Station, Fordham University, 53 Whippoorwill Road, Armonk, NY 10504, USA Abstract Genomic resources are important and attainable for examining evolutionary change in divergent natural populations of nonmodel species. We utilized two next-generation sequencing (NGS) platforms, 454 and SOLiD 5500XL, to assemble low-coverage transcriptomes of the white-footed mouse (Peromyscus leucopus), a widespread and abundant native rodent in eastern North America. We sequenced liver mRNA transcripts from multiple individuals collected from urban populations in New York City and rural populations in undisturbed protected areas nearby and assem- bled a reference transcriptome using 1 080 065 954 SOLiD 5500XL (75 bp) reads and 3 052 640 454 GS FLX + reads. The reference contained 40 908 contigs with a N50 = 1044 bp and a total content of 30.06 Megabases (Mb). Contigs were annotated from Mus musculus (39.96% annotated) Uniprot databases. We identified 104 655 high-quality single nucleotide polymorphisms (SNPs) and 65 single sequence repeats (SSRs) with flanking primers. We also used nor- malized read counts to identify putative gene expression differences in 10 genes between populations. There were 19 contigs significantly differentially expressed in urban populations compared to rural populations, with gene func- tion annotations generally related to the translation and modification of proteins and those involved in immune responses. -
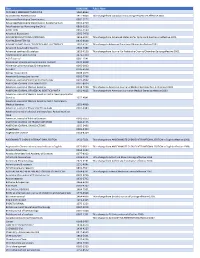
2019 Wiley Title List
Title ISSN/ISBN Public Note ACADEMIC EMERGENCY MEDICINE 1069-6563 ACCOUNTING PERSPECTIVES 1911-382X Title change from Canadian Accounting Perspectives effective 2007. Acta anaesthesiologica Scandinavica 0001-5172 Acta anaesthesiologica Scandinavica. Supplementum 0515-2720 Acta Paediatrica: Nurturing the Child 0803-5253 ADDICTION 0965-2140 Advanced Biosystems 2366-7478 ADVANCED FUNCTIONAL MATERIALS 1616-301X Title change from Advanced Materials for Optics and Electronics effective 2001. ADVANCED MATERIALS 0935-9648 ADVANCED MATERIALS FOR OPTICS AND ELECTRONICS 1057-9257 Title change to Advanced Functional Materials effective 2001. Advanced Sustainable Systems 2366-7486 Advanced synthesis & catalysis 1615-4150 Title change from Journal für Praktische Chemie/Chemiker-Zeitung effective 2001. AEM Education and Training 2472-5390 AIChE Journal 0001-1541 Alcoholism: clinical and experimental research 0145-6008 Alimentary pharmacology & therapeutics 0269-2813 ALLERGY 0105-4538 Allergy. Supplement 0108-1675 American Business Law Journal 0002-7766 American Journal of Community Psychology 0091-0562 AMERICAN JOURNAL OF HEMATOLOGY 0361-8609 American Journal of Medical Genetics 0148-7299 Title change to American Journal of Medical Genetics Part A effeective 2003. AMERICAN JOURNAL OF MEDICAL GENETICS PART A 1552-4825 Title change from American Journal of Medical Genetics effective 2003. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics 1552-4841 American Journal of Medical Genetics Part C: Seminars in Medical Genetics 1552-4868 American Journal of Physical Anthropology 0002-9483 American journal of physical anthropology. Annual meeting issue American Journal of Political Science 0092-5853 AMERICAN JOURNAL OF TRANSPLANTATION 1600-6135 AMERICAN JOURNAL ON ADDICTIONS 1055-0496 Anaesthesia 0003-2409 Angewandte Chemie 0044-8249 ANGEWANDTE CHEMIE INTERNATIONAL EDITION 1433-7851 Title change from ANGEWANDTE CHEMIE INTERNATIONAL EDITION in English effective 1998.