Related Circular RNA Signature
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Ubiquitination Is Not Omnipresent in Myeloid Leukemia Ramesh C
Editorials Ubiquitination is not omnipresent in myeloid leukemia Ramesh C. Nayak1 and Jose A. Cancelas1,2 1Division of Experimental Hematology and Cancer Biology, Cincinnati Children’s Hospital Medical Center and 2Hoxworth Blood Center, University of Cincinnati Academic Health Center, Cincinnati, OH, USA E-mail: JOSE A. CANCELAS - [email protected] / [email protected] doi:10.3324/haematol.2019.224162 hronic myelogenous leukemia (CML) is a clonal tination of target proteins through their cognate E3 ubiq- biphasic hematopoietic disorder most frequently uitin ligases belonging to three different families (RING, Ccaused by the expression of the BCR-ABL fusion HERCT, RING-between-RING or RBR type E3).7 protein. The expression of BCR-ABL fusion protein with The ubiquitin conjugating enzymes including UBE2N constitutive and elevated tyrosine kinase activity is suffi- (UBC13) and UBE2C are over-expressed in a myriad of cient to induce transformation of hematopoietic stem tumors such as breast, pancreas, colon, prostate, lym- cells (HSC) and the development of CML.1 Despite the phoma, and ovarian carcinomas.8 Higher expression of introduction of tyrosine kinase inhibitors (TKI), the dis- UBE2A is associated with poor prognosis of hepatocellu- ease may progress from a manageable chronic phase to a lar cancer.9 In leukemia, bone marrow (BM) cells from clinically challenging blast crisis phase with a poor prog- pediatric acute lymphoblastic patients show higher levels nosis,2 in which myeloid or lymphoid blasts fail to differ- of UBE2Q2 -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -
![UBE2A (HR6A) [GST-Tagged] E2 – Ubiquitin Conjugating Enzyme](https://docslib.b-cdn.net/cover/1509/ube2a-hr6a-gst-tagged-e2-ubiquitin-conjugating-enzyme-1351509.webp)
UBE2A (HR6A) [GST-Tagged] E2 – Ubiquitin Conjugating Enzyme
UBE2A (HR6A) [GST-tagged] E2 – Ubiquitin Conjugating Enzyme Alternate Names: HHR6A, HR6A, RAD6A, UBC2, EC 6.3.2.19, Ubiquitin-conjugating enzyme E2A Cat. No. 62-0001-020 Quantity: 20 µg Lot. No. 1384 Storage: -70˚C FOR RESEARCH USE ONLY NOT FOR USE IN HUMANS CERTIFICATE OF ANALYSIS Background Physical Characteristics The enzymes of the ubiquitylation pathway Species: human Protein Sequence: play a pivotal role in a number of cellular MSPILGYWKIKGLVQPTRLLLEYLEEKYEEH processes including the regulated and tar- Source: E. coli expression LYERDEGDKWRNKKFELGLEFPNLPYYIDGD geted proteosomal degradation of sub- VKLTQSMAIIRYIADKHNMLGGCPKER strate proteins. Three classes of enzymes Quantity: 20 µg AEISMLEGAVLDIRYGVSRIAYSKDFETLKVD are involved in the process of ubiquityla- FLSKLPEMLKMFEDRLCHKTYLNGDHVTHP tion; activating enzymes (E1s), conjugating Concentration: 1 mg/ml DFMLYDALDVVLYMDPMCLDAFPKLVCFK enzymes (E2s) and protein ligases (E3s). KRIEAIPQIDKYLKSSKYIAWPLQGWQATFG UBE2A is a member of the E2 conjugating Formulation: 50 mM HEPES pH 7.5, GGDHPPKSDLEVLFQGPLGSPNSRVDSTPAR enzyme family and cloning of the human 150 mM sodium chloride, 2 mM RRLMRDFKRLQEDPPAGVSGAPSENNIMVW gene was first described by Koken et al. dithiothreitol, 10% glycerol NAVIFGPEGTPFEDGTFKLTIEFTEEYPNKPPT (1991). UBE2A shares 70% identity with VRFVSKMFHPNVYADGSICLDILQNRWSPTYD its yeast homologue but lacks the acidic c- Molecular Weight: ~45 kDa VSSILTSIQSLLDEPNPNSPANSQAAQLYQENK terminal domain. The ring finger proteins REYEKRVSAIVEQSWRDC RAD5 and RAD18 interact with UBE2A and Purity: >98% by InstantBlue™ SDS-PAGE Tag (bold text): N-terminal glutathione-S-transferase (GST) other members of the RAD6 pathway (Ul- Protease cleavage site: PreScission™ (LEVLFQtGP) rich and Jentsch, 2000). Phosphorylation of Stability/Storage: 12 months at -70˚C; UBE2A (regular text): Start bold italics (amino acid UBE2A by CDK1 and 2 increases its activ- aliquot as required residues 2-152) Accession number: NP_003327 ity during the G2/M phase of the cell cycle (Sarcevic et al., 2002). -

Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay
Copyright by John Francis Anderson 2011 THE ROLE OF SACSIN AS A MOLECULAR CHAPERONE by John Francis Anderson, B.S. Dissertation Presented to the Faculty of the Graduate School of The University of Texas Medical Branch in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy The University of Texas Medical Branch May 2011 Dedication To my wife, Erika R. Anderson. Acknowledgements I am indebted to my mentor, Dr. José M. Barral for his investment in my education. Dr. Barral taught me that curiosity drives scientific investigation and rigorous experiments derive new knowledge. This combination of values is rare to find in a mentor and I am extremely grateful for my experience in his lab. I would like to acknowledge Jason J. Chandler and Paige Spencer for technical assistance. I am especially grateful to Dr. Efrain Siller for daily assistance and discussions on all aspects of this project. I would like to thank Dr. Christian Kaiser providing me a practical education on the design and performance biochemical experiments. I acknowledge Dr. Bernard Brais for generously sharing his time and resources; he provided the rare opportunity to visit an ARSACS patient in her home as well as supplied us with SACS knockout mouse brains. My committee members, Dr. Henry F. Epstein, Dr. Andres F. Oberhauser, Dr. George R Jackson, and Dr. Robert O. Fox and Dr. Darren F. Boehning provided invaluable guidance throughout this work for which I am very grateful. I would like to especially thank Dr. Darren F. Boehning and Dr. Henry F. Epstein for being involved in the details of my project from the beginning of my education at UTMB, through sharing reagents and expertise as well as serving on my committee. -

Cracking the Monoubiquitin Code of Genetic Diseases
International Journal of Molecular Sciences Review Cracking the Monoubiquitin Code of Genetic Diseases 1,2, 1,2, 1,2, Raj Nayan Sewduth y, Maria Francesca Baietti y and Anna A. Sablina * 1 VIB-KU Leuven Center for Cancer Biology, VIB, Herestraat 49, 3000 Leuven, Belgium; [email protected] (R.N.S.); [email protected] (M.F.B.) 2 Department of Oncology, KU Leuven, Herestraat 49, 3000 Leuven, Belgium * Correspondence: [email protected] These authors contributed equally to the work. y Received: 7 April 2020; Accepted: 23 April 2020; Published: 25 April 2020 Abstract: Ubiquitination is a versatile and dynamic post-translational modification in which single ubiquitin molecules or polyubiquitin chains are attached to target proteins, giving rise to mono- or poly-ubiquitination, respectively. The majority of research in the ubiquitin field focused on degradative polyubiquitination, whereas more recent studies uncovered the role of single ubiquitin modification in important physiological processes. Monoubiquitination can modulate the stability, subcellular localization, binding properties, and activity of the target proteins. Understanding the function of monoubiquitination in normal physiology and pathology has important therapeutic implications, as alterations in the monoubiquitin pathway are found in a broad range of genetic diseases. This review highlights a link between monoubiquitin signaling and the pathogenesis of genetic disorders. Keywords: ubiquitin system; genetic diseases; ubiquitin ligase; deubiquitinases; monoubiquitin signaling; vesicular trafficking; protein complex formation 1. Introduction Ubiquitination is a reversible post-translational modification process during which the highly conserved 76-aminoacid protein ubiquitin is conjugated to target proteins. Ubiquitin can be conjugated to a protein substrate via distinct mechanisms. -

Mechanism of Ssph1: a Bacterial Effector Ubiquitin Ligase
© Copyright 2018 Matthew J. Cook Mechanism of SspH1: A Bacterial Effector Ubiquitin Ligase Hijacks the Eukaryotic Ubiquitylation Pathway Matthew J. Cook A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy University of Washington 2018 Reading Committee: Peter S Brzovic, Chair Suzanne Hoppins Ning Zheng Program Authorized to Offer Degree: Biochemistry University of Washington Abstract Mechanism of SspH1: A Bacterial Effector Ubiquitin Ligase Hijacks the Eukaryotic Ubiquitylation Pathway Matthew J. Cook Chair of the Supervisory Committee: Peter S Brzovic Department of Biochemistry Bacterial effector proteins promote the pathogenicity of bacteria by interacting with host cell proteins and modulating signaling pathways in the host cell. Some bacterial effectors hijack the eukaryotic ubiquitylation pathway to promote pathogenicity, despite the absence of the pathway in prokaryotes. One family of bacterial effectors, the IpaH-SspH family of E3 ligases, are unrelated in sequence or structure to eukaryotic E3s. This work elucidates key aspects in the structure and mechanism of one member of the IpaH-SspH family from Salmonella typhimurium, SspH1. Biophysical and biochemical methods were used to examine the structure of SspH1 in solution and the interactions between SspH1 and components of the eukaryotic ubiquitylation pathway, with an emphasis on the mechanisms of ubiquitin transfer from the E2 active site to the SspH1 active site, and from the SspH1 active site to substrate. Important results of this work include 1) a reanalysis of existing crystal structures of IpaH-SspH1 E3 domains leading to a new way of characterizing the structural organization of the SspH1 E3 domain, revealing two independent subdomains. -

Comparative Analysis of the Ubiquitin-Proteasome System in Homo Sapiens and Saccharomyces Cerevisiae
Comparative Analysis of the Ubiquitin-proteasome system in Homo sapiens and Saccharomyces cerevisiae Inaugural-Dissertation zur Erlangung des Doktorgrades der Mathematisch-Naturwissenschaftlichen Fakultät der Universität zu Köln vorgelegt von Hartmut Scheel aus Rheinbach Köln, 2005 Berichterstatter: Prof. Dr. R. Jürgen Dohmen Prof. Dr. Thomas Langer Dr. Kay Hofmann Tag der mündlichen Prüfung: 18.07.2005 Zusammenfassung I Zusammenfassung Das Ubiquitin-Proteasom System (UPS) stellt den wichtigsten Abbauweg für intrazelluläre Proteine in eukaryotischen Zellen dar. Das abzubauende Protein wird zunächst über eine Enzym-Kaskade mit einer kovalent gebundenen Ubiquitinkette markiert. Anschließend wird das konjugierte Substrat vom Proteasom erkannt und proteolytisch gespalten. Ubiquitin besitzt eine Reihe von Homologen, die ebenfalls posttranslational an Proteine gekoppelt werden können, wie z.B. SUMO und NEDD8. Die hierbei verwendeten Aktivierungs- und Konjugations-Kaskaden sind vollständig analog zu der des Ubiquitin- Systems. Es ist charakteristisch für das UPS, daß sich die Vielzahl der daran beteiligten Proteine aus nur wenigen Proteinfamilien rekrutiert, die durch gemeinsame, funktionale Homologiedomänen gekennzeichnet sind. Einige dieser funktionalen Domänen sind auch in den Modifikations-Systemen der Ubiquitin-Homologen zu finden, jedoch verfügen diese Systeme zusätzlich über spezifische Domänentypen. Homologiedomänen lassen sich als mathematische Modelle in Form von Domänen- deskriptoren (Profile) beschreiben. Diese Deskriptoren können wiederum dazu verwendet werden, mit Hilfe geeigneter Verfahren eine gegebene Proteinsequenz auf das Vorliegen von entsprechenden Homologiedomänen zu untersuchen. Da die im UPS involvierten Homologie- domänen fast ausschließlich auf dieses System und seine Analoga beschränkt sind, können domänen-spezifische Profile zur Katalogisierung der UPS-relevanten Proteine einer Spezies verwendet werden. Auf dieser Basis können dann die entsprechenden UPS-Repertoires verschiedener Spezies miteinander verglichen werden. -
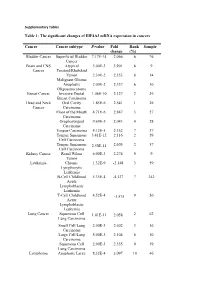
Supplementary Tables
Supplementary Tables Table 1: The significant changes of EIF4A3 mRNA expression in cancers Cancer Cancer subtype P-value Fold Rank Sample change (%) Bladder Cancer Superficial Bladder 7.17E-14 2.066 6 76 Cancer Brain and CNS Atypical 5.00E-3 3.901 6 9 Cancer Teratoid/Rhabdoid Tumor 2.30E-2 2.353 8 14 Malignant Glioma Anaplastic 2.00E-2 2.337 6 10 Oligoastrocytoma Breast Cancer Invasive Ductal 1.06E-10 2.127 2 39 Breast Carcinoma Head and Neck Oral Cavity 1.85E-6 2.541 1 26 Cancer Carcinoma Floor of the Mouth 4.71E-6 2.847 3 27 Carcinoma Oropharyngeal 9.69E-5 2.043 4 28 Carcinoma Tongue Carcinoma 4.12E-5 2.152 7 37 Tongue Squamous 3.41E-12 2.116 2 59 Cell Carcinoma Tongue Squamous 2.50E-11 2.605 2 57 Cell Carcinoma Kidney Cancer Renal Wilms 6.00E-3 2.274 9 9 Tumor Leukemia Chronic 1.32E-9 -2.148 3 59 Lymphocytic Leukemia B-Cell Childhood 5.35E-4 -4.127 7 242 Acute Lymphoblastic Leukemia T-Cell Childhood 4.52E-4 -3.575 9 50 Acute Lymphoblastic Leukemia Lung Cancer Squamous Cell 1.41E-11 2.058 2 62 Lung Carcinoma Small Cell Lung 2.00E-3 2.602 3 10 Carcinoma Large Cell Lung 5.00E-3 2.104 6 10 Carcinoma Squamous Cell 2.00E-3 2.335 9 19 Lung Carcinoma Lymphoma Anaplastic Large 8.32E-4 3.097 10 46 Cell Lymphoma, ALK-Positive Pancreatic Pancreatic Ductal 1.27E-4 2.044 2 14 Cancer Adenocarcinoma Prostate Cancer Prostatic 5.76E-4 2.785 6 34 Intraepithelial Neoplasia Epithelia Prostate Carcinoma 4.37E-4 2.289 7 52 Epithelia Table 2: The association of EIF4A3 expression and the prognosis value in cancer patients CANCER N ENDPOINT HR CORRECTE DATASET -

Protein Homeostasis Networks and the Use of Yeast to Guide Interventions in Alzheimer’S Disease
International Journal of Molecular Sciences Review Protein Homeostasis Networks and the Use of Yeast to Guide Interventions in Alzheimer’s Disease Sudip Dhakal and Ian Macreadie * School of Science, RMIT University, Bundoora, Victoria 3083, Australia; [email protected] * Correspondence: [email protected]; Tel.: +61-3-9925-6627 Received: 29 September 2020; Accepted: 26 October 2020; Published: 28 October 2020 Abstract: Alzheimer’s Disease (AD) is a progressive multifactorial age-related neurodegenerative disorder that causes the majority of deaths due to dementia in the elderly. Although various risk factors have been found to be associated with AD progression, the cause of the disease is still unresolved. The loss of proteostasis is one of the major causes of AD: it is evident by aggregation of misfolded proteins, lipid homeostasis disruption, accumulation of autophagic vesicles, and oxidative damage during the disease progression. Different models have been developed to study AD, one of which is a yeast model. Yeasts are simple unicellular eukaryotic cells that have provided great insights into human cell biology. Various yeast models, including unmodified and genetically modified yeasts, have been established for studying AD and have provided significant amount of information on AD pathology and potential interventions. The conservation of various human biological processes, including signal transduction, energy metabolism, protein homeostasis, stress responses, oxidative phosphorylation, vesicle trafficking, apoptosis, endocytosis, and ageing, renders yeast a fascinating, powerful model for AD. In addition, the easy manipulation of the yeast genome and availability of methods to evaluate yeast cells rapidly in high throughput technological platforms strengthen the rationale of using yeast as a model. -

Gene Expression Changes After Parental Exposure to Metals in the Sea Urchin Affect Timing of Genetic Programme of Embryo Development
biology Article Gene Expression Changes after Parental Exposure to Metals in the Sea Urchin Affect Timing of Genetic Programme of Embryo Development Tiziana Masullo 1,†, Girolama Biondo 2,†, Marilena Di Natale 1,3,†, Marcello Tagliavia 4 , Carmelo Daniele Bennici 1, Marianna Musco 1, Maria Antonietta Ragusa 5, Salvatore Costa 5 , Angela Cuttitta 1,* and Aldo Nicosia 4,* 1 Institute for Studies on the Mediterranean-National Research Council (ISMED-CNR), Detached Unit of Palermo, Via Filippo Parlatore 65, 90145 Palermo, Italy; [email protected] (T.M.); [email protected] (M.D.N.); [email protected] (C.D.B.); [email protected] (M.M.) 2 Institute for Anthropic Impacts and Sustainability in Marine Environment-National Research Council (IAS-CNR), Detached Unit of Capo Granitola, Via del mare 3, 91021 Campobello di Mazara, Italy; [email protected] 3 Department of Earth and Marine Science (DiSTeM), University of Palermo, Via Archirafi 20, 90123 Palermo, Italy 4 Institute for Biomedical Research and Innovation-National Research Council-(IRIB-CNR), Via Ugo La Malfa 153, 90146 Palermo, Italy; [email protected] 5 Department of Biological, Chemical and Pharmaceutical Sciences and Technologies (STEBICEF), University of Palermo, Viale delle Scienze Ed. 16, 90128 Palermo, Italy; [email protected] (M.A.R.); [email protected] (S.C.) * Correspondence: [email protected] (A.C.); [email protected] (A.N.) † These authors contributed equally to this work. Citation: Masullo, T.; Biondo, G.; Natale, M.D.; Tagliavia, M.; Bennici, Simple Summary: Intergenerational and transgenerational effects, in which exposure to stressors C.D.; Musco, M.; Ragusa, M.A.; Costa, in a parental generation affects the phenotype of the offspring have been connected to anthropic S.; Cuttitta, A.; Nicosia, A. -

How Is the Fidelity of Proteins Ensured in Terms of Both Quality and Quantity at the Endoplasmic Reticulum? Mechanistic Insights Into E3 Ubiquitin Ligases
International Journal of Molecular Sciences Review How Is the Fidelity of Proteins Ensured in Terms of Both Quality and Quantity at the Endoplasmic Reticulum? Mechanistic Insights into E3 Ubiquitin Ligases Ji An Kang 1,2 and Young Joo Jeon 1,2,* 1 Department of Biochemistry, College of Medicine, Chungnam National University, Daejeon 35015, Korea; [email protected] 2 Department of Medical Science, College of Medicine, Chungnam National University, Daejeon 35015, Korea * Correspondence: [email protected] Abstract: The endoplasmic reticulum (ER) is an interconnected organelle that plays fundamental roles in the biosynthesis, folding, stabilization, maturation, and trafficking of secretory and transmembrane proteins. It is the largest organelle and critically modulates nearly all aspects of life. Therefore, in the endoplasmic reticulum, an enormous investment of resources, including chaperones and protein folding facilitators, is dedicated to adequate protein maturation and delivery to final destinations. Unfortunately, the folding and assembly of proteins can be quite error-prone, which leads to the generation of misfolded proteins. Notably, protein homeostasis, referred to as proteostasis, is constantly exposed to danger by flows of misfolded proteins and subsequent protein aggregates. To maintain proteostasis, the ER triages and eliminates terminally misfolded proteins by delivering substrates to the ubiquitin–proteasome system (UPS) or to the lysosome, which is termed ER- associated degradation (ERAD) or ER-phagy, respectively. ERAD not only eliminates misfolded or Citation: Kang, J.A.; Jeon, Y.J. How unassembled proteins via protein quality control but also fine-tunes correctly folded proteins via Is the Fidelity of Proteins Ensured in protein quantity control. Intriguingly, the diversity and distinctive nature of E3 ubiquitin ligases Terms of Both Quality and Quantity at the Endoplasmic Reticulum? determine efficiency, complexity, and specificity of ubiquitination during ERAD. -

Insights Into the Mechanism of Action of 13-Cis Retinoic Acid
The Pennsylvania State University The Graduate School College of Medicine INSIGHTS INTO THE MECHANISM OF ACTION OF 13-CIS RETINOIC ACID IN SUPPRESSING SEBACEOUS GLAND FUNCTION A Thesis in Molecular Medicine by Amanda Marie Nelson © 2007 Amanda Marie Nelson Submitted in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy May 2007 The thesis of Amanda Marie Nelson was reviewed and approved* by the following: Diane M. Thiboutot Professor of Dermatology Thesis Advisor Chair of Committee Gary A. Clawson Professor of Pathology, Biochemistry and Molecular Biology Mark Kester Distinguished Professor of Pharmacology Jeffrey M. Peters Associate Professor of Molecular Toxicology Jong K. Yun Assistant Professor of Pharmacology Craig Meyers Professor of Microbiology and Immunology Co-chair, Molecular Medicine Graduate Degree Program *Signatures are on file in the Graduate School iii ABSTRACT Nearly 40-50 million people of all races and ages in the United States have acne, making it the most common skin disease. Although acne is not a serious health threat, severe acne can lead to disfiguring, permanent scarring; increased anxiety; and depression. Isotretinoin (13-cis Retinoic Acid) is the most potent agent that affects each of the pathogenic features of acne: 1) follicular hyperkeratinization, 2) the activity of Propionibacterium acnes, 3) inflammation and 4) increased sebum production. Isotretinoin has been on the market since 1982 and even though it has been prescribed for 25 years, extensive studies into its molecular mechanism of action in human skin and sebaceous glands have not been done. Since isotretinoin is a teratogen, there is a clear need for safe and effective alternative therapeutic agents.