Absolute Configuration by Vibrational Circular Dichroism of Anti
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

II. Stereochemistry 5
B.Sc.(H) Chemistry Semester - II Core Course - III (CC-III) Organic Chemistry - I II. Stereochemistry 5. Physical and Chemical Properties of Stereoisomers Dr. Rajeev Ranjan University Department of Chemistry Dr. Shyama Prasad Mukherjee University, Ranchi 1 Syllabus & Coverage Syllabus II Stereochemistry: Fischer Projection, Newmann and Sawhorse Projection formulae and their interconversions. Geometrical isomerism: cis–trans and syn-anti isomerism, E/Z notations with Cahn Ingold and Prelog (CIP) rules for determining absolute configuration. Optical Isomerism: Optical Activity, Specific Rotation, Chirality/Asymmetry, Enantiomers, Molecules with two or more chiral-centres, Distereoisomers, Meso structures, Racemic mixture. Resolution of Racemic mixtures. Relative and absolute configuration: D/L and R/S designations. Coverage: 1. Types of Isomers : Comparing Structures 2. Optical Activity 3. Racemic Mixtures : Separation of Racemic Mixtures 4. Enantiomeric Excess and Optical Purity 5. Relative and Absolute Configuration 6. Physical and Chemical Properties of Stereoisomers 2 Stereochemistry Types of Isomers Dr. Rajeev Ranjan 3 Stereochemistry Determining the Relationship Between Two Non-Identical Molecules Dr. Rajeev Ranjan 4 Stereochemistry Comparing Structures: Are the structures connected the same? yes no Are they mirror images? Constitutional Isomers yes no Enantiomers Enantiomers Is there a plane of symmetry? All chiral centers will be opposite between them. yes no Meso Diastereomers superimposable Dr. Rajeev Ranjan 5 Stereochemistry Optical Activity: • The chemical and physical properties of two enantiomers are identical except in their interaction with chiral substances. • The physical property that differs is the behavior when subjected to plane-polarized light ( this physical property is often called an optical property). • Plane-polarized (polarized) light is light that has an electric vector that oscillates in a single plane. -

Enantiomeric Resolution and Absolute Configuration of a Chiral Δ-Lactam
molecules Article Enantiomeric Resolution and Absolute Configuration of a Chiral δ-Lactam, Useful Intermediate for the Synthesis of Bioactive Compounds 1, 1, 1 1 Roberta Listro y, Giacomo Rossino y, Serena Della Volpe , Rita Stabile , Massimo Boiocchi 2 , Lorenzo Malavasi 3, Daniela Rossi 1,* and Simona Collina 1 1 Department of Drug Sciences, University of Pavia, via Taramelli 12, 27100 Pavia, Italy; [email protected] (R.L.); [email protected] (G.R.); [email protected] (S.D.V.); [email protected] (R.S.); [email protected] (S.C.) 2 Centro Grandi Strumenti, University of Pavia, via Bassi 21, 27100 Pavia, Italy; [email protected] 3 Department of Chemistry, University of Pavia, via Taramelli 12, 27100 Pavia, Italy; [email protected] * Correspondence: [email protected] These authors contributed equally to this work. y Academic Editor: Józef Drabowicz Received: 2 December 2020; Accepted: 18 December 2020; Published: 19 December 2020 Abstract: During the past several years, the frequency of discovery of new molecular entities based on γ- or δ-lactam scaffolds has increased continuously. Most of them are characterized by the presence of at least one chiral center. Herein, we present the preparation, isolation and the absolute configuration assignment of enantiomeric 2-(4-bromophenyl)-1-isobutyl-6-oxopiperidin-3-carboxylic acid (trans-1). For the preparation of racemic trans-1, the Castagnoli-Cushman reaction was employed. (Semi)-preparative enantioselective HPLC allowed to obtain enantiomerically pure trans-1 whose absolute configuration was assigned by X-ray diffractometry. Compound (+)-(2R,3R)-1 represents a reference compound for the configurational study of structurally related lactams. -

Absolute Configuration Determination
Chirality in Pharmaceutical Compounds and Absolute Configuration Determination Crystallization: Screening, Application & Process Development X-ray Diffraction Unit Jordi Benet-Buchholz Barcelona, 11 April 2019 Institut Català d’Investigació Química Av. Països Catalans 16 1 43007 Tarragona (Spain) From chiral crystals to the absolute configuration of molecules: Is it an easy way? (–)-Galiellalactone *S OH R * *S O O *R Orthorhombic , P 2 2 2 Absolute configuration: 1 1 1 R1: 3,45 % 4S, 5aR, 7aR, 7bS Flack: 0.01(19) Hooft/Parsons: -0.06(04)/-0.04(05) • F. Nussbaum, R. Hanke, T. Fahrig, J. Benet-Buchholz; Eur. J. Org. Chem. 2004, 2783-2790. 2 Determination of Absolute Configuration of APIs Definitions in Chirality Precedents Methodologies Single Crystal X-ray Structure Determination: Background Candidate samples Sample and Crystal selection Validation and measurements Examples Final schedule 3 Chirality Definitions in Chirality: An object or a system is chiral if it is distinguishable from its mirror image; that is, it cannot be superposed onto it. The word chirality is derived from the Greek word meaning “hand” Paula Benet The most universally recognized example for chirality are the human hands. Live is chiral 4 Chirality Definitions in Chirality: - Each molecule which cannot be superposed with its mirror image is “chiral”. - Molecules with a mirror plane are called “achiral”. The feature that is most often the cause of chirality in molecules is the presence of an asymmetric carbon atom (four different substituents) Two mirror images of a chiral molecule are called enantiomers or optical isomers Foto cristall papallona 5 Conglomerate of crystals forming enantiomeric butterflies Chirality Definitions in Chirality: Pairs of enantiomers are often designated as “right- or left-handed”. -

Absolute Configuration Some Definitions
Absolute Structure Absolute Configuration Some definitions • Absolute Configuration -> spatial arrangement of the atoms for a chiral molecule (R/S, P/M or D/L assignment). • Absolute Structure -> spatial arrangement of atoms in a noncentrosymmetric crystal structure (unit-cell, space group) • Chiral molecules Molecules that cannot be superimposed with their mirror image • Two mirror images of a chiral molecule are called enantiomers, they are optical isomers • Determination of absolute configuration -> handness of the molecule Origin of chirality • Asymmetric carbon atoms (R/S) • Axial chirality R-Binol S-Binol • Chiral Propeller Arrangement (P/M or / ) Helicenes In the solid state • Chiral molecules can crystallize as an enantiopure bulk sample or as a racemic mixture. • For enantiopure crystals Space group restriction: Only 65 space groups allowed for chiral molecules: No Inversion Center / No Mirror / No Glide Plane These include 11 pairs of eniantomorph space groups (screw axes of opposite handedness) eg: P41/P43 or P61/P65 Space Group Restrictions 85 % Racemic mixture In the solid state • 1) Conglomerate: a mixture of well-resolved crystals of both enantiomers Chiral space group Individual crystals have an optical activity 5-10 % • 2) Racemates: One type of crystal containing the two enantiommers in a well defined stoechiometry. No optical activity Usually centrosymmetric space group 90-95 % Conglomerate Racemates Racemic mixture In the solid state • 3) Inversion twin : twinned crystals of both enantiomers Chiral space group • 4) -

1 Fundamentals of the Stereochemistry of Organophosphorus Compounds
1 1 Fundamentals of the Stereochemistry of Organophosphorus Compounds 1.1 Historical Background Natural processes are subordinate to geometrodynamics – the theory describing physical objects, geometrical spacetime, and associated phenomena completely in terms of geometry, and her elder sister – symmetry. Symmetry/asymmetry is one of the basic concepts in modern natural science [1]. Research into this field began in the Middle Ages, when the birefringent properties of calcite were discovered. In 1669, Bartholinus observed the double refractive properties of the calcite Iceland spar. Later, in 1801, the mineralogist Haui found that quartz crystals are enantiomorphic, representing mirror images of one another. In 1815, another French naturalist J.-B. Biot discovered that certain chemical compounds rotate the plane of a beam of polarized light [2]. Biot constructed the first polarimeter and he also discovered that many natural compounds exhibit optical activity, that is, they rotate the plane of circularly polarized light. Studying crystals under a microscope, Biot discovered two types of crystals. The sample consisting of crystals of one type turned polarized light clockwise and that from another type in the opposite direction. A mixture of the two types of crystals had a neutral effect on polar- ized light. The nature of this property remained a mystery until 1848, when Louis Pasteur proposed that it had a molecular basis originating from some form of dissymmetry [3]. Pasteur separated the left and right hemihedral crystals of the sodium-ammonium salt of D,L-tartaric acid under a microscope, and connected the opposite optical activity to the mirror image of these crystals. Pasteur termed the mixture creating polarization as dissymetric and the phenomenon as dissymmetry (asymmetry). -

Distinction of Enantiomers by NMR
REVIEWS Disinction of enantiomers by NMR spectroscopy using chiral orienting media Burkhard Luy Abstract | NMR spectroscopy is a very important analytical tool in modern organic and inorganic chemistry. Next to the identification of molecules and their structure determination, it is also used for the distinction of enantiomers and the measurement of enantiomeric purity. This article gives a brief review of the techniques being developed for enantiomeric differentiation by virtue of chiral alignment media and their induction of enantiomerically dependent anisotropic NMR parameters like residual dipolar couplings. An overview of existing chiral alignment media, a brief introduction into the basic theory and measurement of the various anisotropic parameters, and several example applications are given. 1. Introduction valuable compounds one doesn’t necessarily want to NMR spectroscopy is one of the most important irreversibly modify the substance. analytical tools in modern organic and inorganic Another possibility for the distinction of chemistry as it is the only tool that allows the enantiomers is the orientation of the molecule of determination of molecular structures at atomic interest in a so-called chiral alignment medium. resolution in solution. It is used to identify the In this case, the molecule is partially aligned constitution, conformation and configuration and anisotropic NMR parameters like residual of countless molecules every day. However, the quadrupolar couplings, residual dipolar couplings magnetic field used for the induction of the Zeeman and residual chemical shift anisotropy can be splitting is per se achiral so that enantiomers measured2–4. As the orientation in a chiral have identical properties and therefore identical alignment medium is different for the two NMR spectra. -
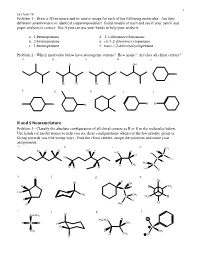
R and S Nomemclature Problem 3 - Classify the Absolute Configuration of All Chiral Centers As R Or S in the Molecules Below
1 Lecture 16 Problem 1 - Draw a 3D structure and its mirror image for each of the following molecules. Are they different (enantiomers) or identical (superimposable)? Build models of each and see if your pencil and paper analysis is correct. See if you can use your hands to help your analysis. a. 1-bromopentane d. 1,1-dibromocyclopentane b. 2-bromopentane e. cis-1,2-dibromocyclopentane c. 3-bromopentane f. trans-1,2-dibromocyclopentane Problem 2 - Which molecules below have stereogenic centers? How many? Are they all chiral centers? a. b. c. d. e. Cl OH Br Br Cl Br Br f. g. h. i. j. Br Br Br Br R and S Nomemclature Problem 3 - Classify the absolute configuration of all chiral centers as R or S in the molecules below. Use hands (or model atoms) to help you see these configurations whenever the low priority group is facing towards you (the wrong way). Find the chiral centers, assign the priorities and make your assignments. a. b. c. d. CH3 Cl H H H3C H H H H3C Br Cl C HO C CH3 I H H3C H CH3 C H H H H3C H e. O f. g. h. H Br Br OH H H H CH3 C H3C CH3 CH3 C H3C H H H H C H Cl 3 i. j. k. l. Cl CH2CH3 Br H3CH2C H C Br H CH3 Cl S CH3 D CH3 H CH3 O H 2 Lecture 16 Pi Bond Priority Problem 4 - Evaluate the order of priority in each part. -
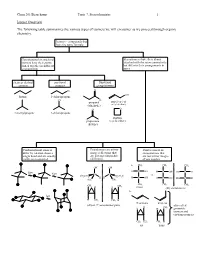
Isomer Overview the Following Table Summarizes the Various Types Of
Chem 201/Beauchamp Topic 7, Stereochemistry 1 Isomer Overview The following table summarizes the various types of isomers we will encounter as we proceed through organic chemistry. Isomers - compounds that have the same formula Constitutional or structural Stereoisomers have their atoms isomers have their atoms attached with the same connectivity, joined together in different but differ in their arrangements in arrangements space chain or skeletal positional functional isomers isomers group isomers Cl O OH butane 1-chloropropane Cl H propanal prop-2-en-1-ol (aldehyde) (allyl alcohol) O O 2-methylpropane 2-chloropropane oxatane propanone (cyclic ether) (ketone) Conformational isomers Enantiomers are mirror Diastereomers are differ by rotation about a image reflections that stereoisomers that single bond and are usually are not superimposable are not mirror images easily interconverted. (different). of one another. CH a. 3 CH3 CH3 OH OH fast H C OH H C OH HO C H fast C C vs. CH3CH2 H2CH3C H H H C OH H C OH HO C H H3C CH3 CH 3 CH3 CH3 CH3 CH H H 3 meso (dl) enantiomers b. fast H CH3 CH3 H E or trans Z or cis (dl) or () enantiomer pairs also called H CH H H 3 geometric isomers and cis/trans isomers CH3 CH3 CH3 H cis trans Chem 201/Beauchamp Topic 7, Stereochemistry 2 Essential Vocabulary of Stereochemistry – terms to know for this topic Chiral center – a tetrahedral atom with four different groups attached, a single chiral center is not superimposable on its mirror image, also called a stereogenic center, and asymmetric center Stereogenic center – any center where interchange of two groups produces stereoisomers (different structures), could be a chiral center producing R/S differences, or an alkene producing E/Z differences or cis/trans in a ring Absolute configuration (R and S) – the specific 3 dimensional configuration about a tetrahedral shape, determined by assigning priorities (1-4) based on the highest atomic number of an atom at the first point of difference. -

Chapter 5: Stereoisomerism- Three-Dimensional Arrangement of Atoms (Groups) in Space 5.1 Overview of Isomerism
Chapter 5: Stereoisomerism- three-dimensional arrangement of atoms (groups) in space 5.1 Overview of Isomerism Isomers: different chemical compounds with the same formula Constitutional isomers: same formula, but different connectivity of atoms (or groups) C5H12 C4H10O C4H11N NH O 2 OH NH2 butanol diethyl ether different carbon skeleton different functional group different position of FG Stereoisomers: molecules with the same connectivity but different spatial arrangement of atoms (groups) H C CH 3 3 H3C H H H H CH3 cis-1,2-dimethylcyclopropane trans-1,2-dimethylcyclopropane H H H CH3 H C CH H C H 3 3 3 93 cis-2-butene trans-2-butene 5.2 Introduction to Stereoisomerism Enantiomers: non-superimposable mirror image isomers. Enantiomers are related to each other much like a right hand is related to a left hand Enantiomers have identical physical properties, e.g., bp, mp, etc. Chirality (derived from the Greek word for hand). Enantiomers are said to be chiral. 94 47 Chirality Center: Chiral molecules most commonly contain a carbon with four different groups; the carbon is referred to as a chiral center, or asymmetric center, or stereogenic center, or stereocenter. H H HO OH CO2H HO2C H3C CH3 H H H CO2H HO HO CO2H CO2H H3C H3C H3C HO CO2H H HO H3C H H H OH HO HO CO2H CO2H H3C H3C HO H CH3 CO2H HO2C H3C 95 5.3 Designating Configuration Using the Cahn-Ingold-Prelog System – Assigning the Absolute Configuration 1a. Look at the four atoms directly attached to the chiral carbon atom and rank them according to decreasing atomic number. -

7.3 Designating Configuration of Enantiomers
Hornback_Ch07_219-256 12/14/04 7:00 PM Page 224 224 CHAPTER 7 I STEREOCHEMISTRY II PROBLEM 7.3 Indicate whether each of these objects or molecules has a plane of symmetry: a) Idealized human face b) Pencil c) Ear Cl CH3 O d) e) f) H3CCH3 O Cl± Br± CH g) C 3 h) C CH3 ± H H Cl Cl ± 7.3 Designating Configuration of Enantiomers Suppose we are working with one enantiomer of a chiral compound. How can we indi- cate which enantiomer is being used when writing about it? Or how can we look up the properties of this enantiomer in a reference book? We need a method to designate the configuration of the enantiomer, to denote the three-dimensional arrangement of the four groups around the chirality center, other than drawing the structure. In the case of many everyday chiral objects, the terms right and left are used, as in a left shoe or right- handed golf clubs. In Section 6.2 we learned how to designate the configuration of geo- metrical isomers using the Cahn-Ingold-Prelog sequence rules and the labels Z and E. The method to designate the configuration of enantiomers uses these same rules. The following steps are used to assign the configuration of a chiral compound: STEP 1 Assign priorities from 1 through 4 to the four groups bonded to the chirality center using the Cahn-Ingold-Prelog sequence rules presented in Section 6.2.The group with the highest priority receives number 1, and the lowest-priority group receives number 4. -
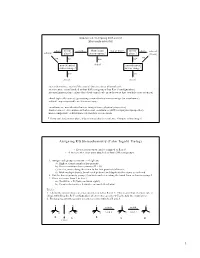
Cahn–Ingold–Prelog)
Handout #6, 5.12 Spring 2003, 2/28/03 Stereochemistry yes mirror zero How many two or more mirror yes achiral achiral plane? stereocenters? plane? (meso) no no no one no* non-identical chiral non-identical mirror image? mirror image? yes yes chiral chiral stereochemistry: study of the spatial characteristics of a molecule stereocenter: atom bonded to four different groups (has R or S configuration) internal mirror plane: plane that divides molecule in such a way that two halves are identical chiral (optically active): possessing a non-identical mirror image (an enantiomer) achiral: superimposable on its mirror image enantiomers: non-identical mirror images (same physical properties) diastereomers: stereoisomers that are not enantiomers (different physical properties) meso compound: achiral molecule that has stereocenters * If you can't find a mirror plane, it doesn't mean that there isn't one. Compare mirror images! Assigning R/S Stereochemistry (Cahn–Ingold–Prelog) • Every stereocenter can be assigned as R or S. • A stereocenter is an atom attached to four different groups. 1. Assign each group a priority (1 = highest). a) Highest atomic number has priority. b) Heavier isotopes have priority (D > H). c) In a tie, move along the chain to the first point of difference. d) With multiple bonds, break each pi-bond and duplicate the atoms at each end. 2. Put the lowest priority group (4) in back and view along the bond from carbon to group 4. 3. Draw an arrow from 1 to 2 to 3. a) Clockwise = R (Your car turns right!) b) Counterclockwise = S (sinister means left in Latin) Tricks: 1. -

Basic Organic Stereochemistry
BASIC ORGANIC STEREOCHEMISTRY ERNEST L. ELIEL SAMUEL H. WILEN MICHAEL P. DOYLE Preface xiii 1 Introduction 1 1-1. Scope, 1 1-2. History, 1 1-3. Polarimetry and Optical Rotation, 5 References, 7 2 Structure 8 2-1 . Meaning, Factorization, Internal Coordinates, Isomers, 8 2-2. Constitution, 1 1 2-3. Configuration, 1 3 2-4. Conformation, 1 5 2-5 . Determination of Structure, 1 7 2-6. A Priori Calculation of Structure, 20 2-7 . Molecular Models, 25 References, 26 3 Stereoisomers 30 3-1. Nature of Stereoisomers, 3 0 a. General, 30 b. Barriers Between Stereoisomers and Residual Stereoisomers, 3 4 3-2. Enantiomers, 3 6 3-3. Diastereomers, 3 9 a. General Cases, 39 b . Degenerate Cases, 42 References, 44 4 Symmetry 45 4-1 . Introduction, 4 5 4-2. Symmetry Elements, 45 4-3. Symmetry Operators and Symmetry Point Groups, 48 a. Point Groups Containing Chiral Molecules, 4 9 b. Point Groups Containing Only Achiral Molecules, 5 1 4-4. Averaged Symmetry, 5 8 4-5. Symmetry and Molecular Properties, 59 a. Rotation of Polarized Light, 5 9 b. Dipole Moment, 6 1 c. Symmetry Number, 6 2 References, 63 5 Configuration 65 5-1 . Definitions : Relative and Absolute Configuration, 6 5 5-2. Absolute Configuration and Notation, 67 5-3. Determination of Absolute Configuration, 7 5 a. Bijvoet Method, 75 b. Theoretical Approaches, 77 c. Modification of Crystal Morphology in the Presence of Additives, 7 7 5-4. Relative Configuration and Notation, 79 5-5. Determination of Relative Configuration of Saturated Aliphati c Compounds, 8 4 a.