Differential Cerna Expression and Interaction Analysis in Coronary Artery Disease Sheng Kang1*#, Yong Ye1#, Guang Xia1, Haibo Liu1,2* 1
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Generation of Knock-In Primary Human T Cells Using Cas9 Ribonucleoproteins
Generation of knock-in primary human T cells using Cas9 ribonucleoproteins Kathrin Schumanna,b,1, Steven Linc,1, Eric Boyera,b, Dimitre R. Simeonova,b,d, Meena Subramaniame,f, Rachel E. Gatee,f, Genevieve E. Haliburtona,b, Chun J. Yee, Jeffrey A. Bluestonea, Jennifer A. Doudnac,g,h,i,j,2, and Alexander Marsona,b,g,2 aDiabetes Center, University of California, San Francisco, CA 94143; bDivision of Infectious Diseases, Department of Medicine, University of California, San Francisco, CA 94143; cDepartment of Molecular and Cell Biology, University of California, Berkeley, CA 94720; dBiomedical Sciences Graduate Program, University of California, San Francisco, CA 94143; eDepartment of Epidemiology and Biostatistics, Department of Bioengineering and Therapeutic Sciences, Institute for Human Genetics, University of California, San Francisco, CA 94143; fBiological and Medical Informatics Graduate Program, University of California, San Francisco, CA 94158; gInnovative Genomics Initiative, University of California, Berkeley, CA 94720; hHoward Hughes Medical Institute, University of California, Berkeley, CA 94720; iDepartment of Chemistry, University of California, Berkeley, CA 94720; and jPhysical Biosciences Division, Lawrence Berkeley National Laboratory, Berkeley, CA 94720 Contributed by Jennifer A. Doudna, June 29, 2015 (sent for review January 23, 2015) T-cell genome engineering holds great promise for cell-based therapies with specific replacement sequence, rather than deleted (14). Effi- for cancer, HIV, primary immune deficiencies, and autoimmune dis- cient technology to promote homologous recombination in T cells eases, but genetic manipulation of human T cells has been challenging. could eventually allow therapeutic correction of mutations that affect Improved tools are needed to efficiently “knock out” genes and “knock specialized T-cell functions. -

Small Nucleolar Rnas Determine Resistance to Doxorubicin in Human Osteosarcoma
International Journal of Molecular Sciences Article Small Nucleolar RNAs Determine Resistance to Doxorubicin in Human Osteosarcoma Martina Godel 1, Deborah Morena 1, Preeta Ananthanarayanan 1, Ilaria Buondonno 1, Giulio Ferrero 2,3 , Claudia M. Hattinger 4, Federica Di Nicolantonio 1,5 , Massimo Serra 4 , 1 2 1, , 1, , Riccardo Taulli , Francesca Cordero , Chiara Riganti * y and Joanna Kopecka * y 1 Department of Oncology, University of Torino, 1026 Torino, Italy; [email protected] (M.G.); [email protected] (D.M.); [email protected] (P.A.); [email protected] (I.B.); [email protected] (F.D.N.); [email protected] (R.T.) 2 Department of Computer Science, University of Torino, 10149 Torino, Italy; [email protected] (G.F.); [email protected] (F.C.) 3 Department of Clinical and Biological Sciences, University of Torino, 10043 Orbassano, Italy 4 Laboratory of Experimental Oncology, Pharmacogenomics and Pharmacogenetics Research Unit, IRCCS Istituto Ortopedico Rizzoli, 40136 Bologna, Italy; [email protected] (C.M.H.); [email protected] (M.S.) 5 Candiolo Cancer Institute, FPO–IRCCS, 10060 Candiolo, Italy * Correspondence: [email protected] (C.R.); [email protected] (J.K.); Tel.: +39-0116705857 (C.R.); +39-0116705849 (J.K.) These authors equally contributed to this work. y Received: 31 May 2020; Accepted: 21 June 2020; Published: 24 June 2020 Abstract: Doxorubicin (Dox) is one of the most important first-line drugs used in osteosarcoma therapy. Multiple and not fully clarified mechanisms, however, determine resistance to Dox. With the aim of identifying new markers associated with Dox-resistance, we found a global up-regulation of small nucleolar RNAs (snoRNAs) in human Dox-resistant osteosarcoma cells. -

Node and Edge Attributes
Cytoscape User Manual Table of Contents Cytoscape User Manual ........................................................................................................ 3 Introduction ...................................................................................................................... 48 Development .............................................................................................................. 4 License ...................................................................................................................... 4 What’s New in 2.7 ....................................................................................................... 4 Please Cite Cytoscape! ................................................................................................. 7 Launching Cytoscape ........................................................................................................... 8 System requirements .................................................................................................... 8 Getting Started .......................................................................................................... 56 Quick Tour of Cytoscape ..................................................................................................... 12 The Menus ............................................................................................................... 15 Network Management ................................................................................................. 18 The -

Copy Number Variations of Chromosome 16P13.1 Region
Molecular Psychiatry (2011) 16, 17–25 & 2011 Macmillan Publishers Limited All rights reserved 1359-4184/11 www.nature.com/mp ORIGINAL ARTICLE Copy number variations of chromosome 16p13.1 region associated with schizophrenia A Ingason1,2, D Rujescu3, S Cichon4,5, E Sigurdsson6, T Sigmundsson6, OPH Pietila¨inen7, JE Buizer-Voskamp8,9, E Strengman9, C Francks10, P Muglia10, A Gylfason1, O Gustafsson1, PI Olason1, S Steinberg1, T Hansen2, KD Jakobsen2, HB Rasmussen2, I Giegling3, H-J Mo¨ller3, A Hartmann3, C Crombie11, G Fraser11, N Walker12, J Lonnqvist13, J Suvisaari13, A Tuulio-Henriksson13, E Bramon14, LA Kiemeney15, B Franke16, R Murray14, E Vassos14, T Toulopoulou14,TWMu¨hleisen4, S Tosato17, M Ruggeri17, S Djurovic18,19, OA Andreassen18,19, Z Zhang20, T Werge2, RA Ophoff9,21, GROUP Investigators22, M Rietschel23,MMNo¨then4,5, H Petursson6, H Stefansson1, L Peltonen7,24,25, D Collier14, K Stefansson1 and DM St Clair11 1deCODE genetics, Reykjavı´k, Iceland; 2Research Institute of Biological Psychiatry, Mental Health Centre Sct. Hans, Copenhagen University Hospital, Roskilde, Denmark; 3Division of Molecular and Clinical Neurobiology, Department of Psychiatry, Ludwig-Maximilians-University and Genetics Research Centre GmbH, Munich, Germany; 4Department of Genomics, Life & Brain Center, University of Bonn, Bonn, Germany; 5Institute of Human Genetics, University of Bonn, Bonn, Germany; 6Department of Psychiatry, National University Hospital, Reykjavı´k, Iceland; 7Department for Molecular Medicine, National Public Health Institute, Helsinki, -

Epigenetic Modifications by Polyphenolic Compounds Alter Gene
© 2018. Published by The Company of Biologists Ltd | Biology Open (2018) 7, bio035196. doi:10.1242/bio.035196 RESEARCH ARTICLE Epigenetic modifications by polyphenolic compounds alter gene expression in the hippocampus Tal Frolinger1,*, Francis Herman1,*, Ali Sharma1, Steven Sims1, Jun Wang1 and Giulio Maria Pasinetti1,2,‡ ABSTRACT methylcytosine dioxygenases (TETs). These epigenetic In this study, we developed an experimental protocol leveraging modifications are known to regulate gene expression in a region enhanced reduced representation bisulphite sequencing to specific manner. Methylation of cytosine residues found in gene investigate methylation and gene expression patterns in the promoter regions is associated with suppression of gene expression hippocampus in response to polyphenolic compounds. We report (Schübeler, 2015). However, evidence to date has yet to establish a that the administration of a standardized bioavailable polyphenolic consistent relationship between the methylation of intronic, exonic, preparation (BDPP) differentially influences methylated cytosine or untranslated regions (UTR) and the expression pattern of the ’ patterns in introns, UTR and exons in hippocampal genes. We gene s corresponding proteins. subsequently established that dietary BDPP-mediated changes in Previous studies have established that dietary polyphenols alter methylation influenced the transcriptional pattern of select genes that the epigenetic characteristics of DNA by regulating the enzymatic DNMTs are involved in synaptic plasticity. In addition, we showed dietary activity of (Paluszczak et al., 2010) and histone BDPP mediated changes in the transcriptional pattern of genes deacetylases (Chung et al., 2010). For example, recent evidence associated with epigenetic modifications, including members of the suggests that bioavailable metabolites derived from dietary BDPP, DNA methyl transferase family (DNMTs) and the Ten-eleven such as malvidin glucoside (Mal-Gluc), decrease the expression of translocation methylcytosine dioxygenases family (TETs). -

Predicting Human Genes Susceptible to Genomic Instability Associated with Alu/Alu-Mediated Rearrangements
Downloaded from genome.cshlp.org on October 1, 2021 - Published by Cold Spring Harbor Laboratory Press Predicting human genes susceptible to genomic instability associated with Alu/Alu-mediated rearrangements Key words: genome instability, Alu, genomic rearrangement, MMBIR, bioinformatics, machine- learning Xiaofei Song1, Christine R. Beck1, Renqian Du1, Ian M. Campbell1, Zeynep Coban-Akdemir1, Shen Gu1, Amy M. Breman1,2, Pawel Stankiewicz1,2, Grzegorz Ira1, Chad A. Shaw1,2, James R. Lupski1,3,4,5* 1. Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX 77030, USA 2. Baylor Genetics, Houston, TX 77021, USA 3. Department of Pediatrics, Baylor College of Medicine, Houston, TX 77030, USA 4. Human Genome Sequencing Center, Baylor College of Medicine, Houston, TX 77030, USA 5. Texas Children’s Hospital, Houston, TX 77030, USA *Corresponding author: James R. Lupski, M.D., Ph.D., D.Sc. (hon), FAAP, FACMG, FANA, FAAAS, FAAS Cullen Professor, Department of Molecular and Human Genetics and Professor of Pediatrics Member, Human Genome Sequencing Center (HGSC) Baylor College of Medicine One Baylor Plaza Houston, Texas 77030 E-mail: [email protected] Phone: (713) 798-6530 Fax: (713) 798-5073 Downloaded from genome.cshlp.org on October 1, 2021 - Published by Cold Spring Harbor Laboratory Press Abstract Alu elements, the short interspersed element numbering >1 million copies per human genome, can mediate the formation of copy number variants (CNVs) between substrate pairs. These Alu/Alu-mediated rearrangements (AAMR) can result in pathogenic variants that cause diseases. To investigate the impact of AAMR on gene variation and human health, we first characterized Alus that are involved in mediating CNVs (CNV-Alus) and observed that these Alus tend to be evolutionarily younger. -
Supplemental Material.Pdf
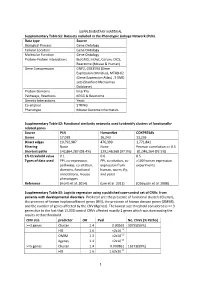
SUPPLEMENTARY MATERIAL Supplementary Table S1: Datasets included in the Phenotypic Linkage Network (PLN). Data type Source Biological Process Gene Ontology Cellular Location Gene Ontology Molecular Function Gene Ontology Protein-Protein Interactions BioGRID, IntAct, Corum, DICS, Reactome (Mouse & Human) Gene Coexpression GNF2, GSE3594 (Gene Expression Omnibus), MTAB-62 (Gene Expression Atlas) , 5 SMD sets (Stanford Microarray Database) Protein Domains InterPro Pathways, Reactions KEGG & Reactome Genetic Interactions Yeast Co-citation STRING Phenotype Mouse Genome Informatics Supplementary Table S2: Functional similarity networks used to identify clusters of functionally- related genes. Source PLN HumanNet COXPRESdb Genes 17,039 16,243 13,236 Direct edges 10,792,987 476,399 1,771,841 Filtering None None Pearson correlation >= 0.5 Shortest paths 142,864,287 (98.4%) 129,146,568 (97.9%) 81,046,264 (92.5%) 1% threshold value 0.1 0.6 0.5 Types of data used PPI, co-expression, PPI, co-citation, co- >100 human expression pathways, co-citation, expression from experiments domains, functional human, worm, fly, annotations, mouse and yeast phenotypes Reference (Honti et al. 2014) (Lee et al. 2011) (Obayashi et al. 2008) Supplementary Table S3: Logistic regression using a published case-control set of CNVs from patients with developmental disorders. Predictor are: the presence of functional clusters (Cluster), the presence of known haploinsufficient genes (HIS), the presence of known disease genes (OMIM), and the number of genes affected by the CNV (#genes). The lowest size threshold considered is >= 3 genes due to the fact that 15,000 control CNVs affected exactly 2 genes which was dominating the results at that threshold. -

Chromosomal Contacts Connect Loci Associated with Autism, BMI and Head Circumference Phenotypes
OPEN Molecular Psychiatry (2017) 22, 836–849 www.nature.com/mp ORIGINAL ARTICLE Chromosomal contacts connect loci associated with autism, BMI and head circumference phenotypes MN Loviglio1,24, M Leleu2,3,24, K Männik1,4, M Passeggeri5, G Giannuzzi1, I van der Werf1, SM Waszak2,3,6, M Zazhytska1, I Roberts-Caldeira5, N Gheldof1, E Migliavacca1,2, AA Alfaiz1,2, L Hippolyte5, AM Maillard5, 2p15 Consortium25, 16p11.2 Consortium26, A Van Dijck7, RF Kooy7, D Sanlaville8, JA Rosenfeld9,10, LG Shaffer11, J Andrieux12, C Marshall13, SW Scherer14,15, Y Shen16,17,18, JF Gusella19,20, U Thorsteinsdottir21, G Thorleifsson21, ET Dermitzakis2,22, B Deplancke2,3, JS Beckmann2,5,23, J Rougemont2,3, S Jacquemont5,27,28 and A Reymond1,28 Copy number variants (CNVs) are major contributors to genomic imbalance disorders. Phenotyping of 137 unrelated deletion and reciprocal duplication carriers of the distal 16p11.2 220 kb BP2-BP3 interval showed that these rearrangements are associated with autism spectrum disorders and mirror phenotypes of obesity/underweight and macrocephaly/microcephaly. Such phenotypes were previously associated with rearrangements of the non-overlapping proximal 16p11.2 600 kb BP4-BP5 interval. These two CNV- prone regions at 16p11.2 are reciprocally engaged in complex chromatin looping, as successfully confirmed by 4C-seq, fluorescence in situ hybridization and Hi-C, as well as coordinated expression and regulation of encompassed genes. We observed that genes differentially expressed in 16p11.2 BP4-BP5 CNV carriers are concomitantly modified in their chromatin interactions, suggesting that disruption of chromatin interplays could participate in the observed phenotypes. We also identified cis- and trans-acting chromatin contacts to other genomic regions previously associated with analogous phenotypes. -
Identification of a Candidate Gene Set Signature for the Risk Of
cancers Article Identification of a Candidate Gene Set Signature for the Risk of Progression in IgM MGUS to Smoldering/Symptomatic Waldenström Macroglobulinemia (WM) by a Comparative Transcriptome Analysis of B Cells and Plasma Cells Alessandra Trojani 1,* , Barbara Di Camillo 2, Luca Emanuele Bossi 1, Livia Leuzzi 1, Antonino Greco 3 , Alessandra Tedeschi 1, Anna Maria Frustaci 1, Marina Deodato 1, Giulia Zamprogna 1, Alessandro Beghini 4 and Roberto Cairoli 1 1 Department of Hematology, ASST Grande Ospedale Metropolitano Niguarda, 20162 Milan, Italy; [email protected] (L.E.B.); [email protected] (L.L.); [email protected] (A.T.); [email protected] (A.M.F.); [email protected] (M.D.); [email protected] (G.Z.); [email protected] (R.C.) 2 Department of Information Engineering, University of Padova, 35131 Padova, Italy; [email protected] 3 Citation: Trojani, A.; Di Camillo, B.; Division of Hematology and Bone Marrow Transplant, Cardinale Giovanni Panico Hospital, Tricase, 73030 Lecce, Italy; [email protected] Bossi, L.E.; Leuzzi, L.; Greco, A.; 4 Department of Health Sciences, University of Milan, 20142 Milan, Italy; [email protected] Tedeschi, A.; Frustaci, A.M.; Deodato, * Correspondence: [email protected]; Tel.: +39-2-64442711 M.; Zamprogna, G.; Beghini, A.; et al. Identification of a Candidate Gene Set Simple Summary: IgM monoclonal gammopathy of undetermined significance (IgM MGUS) is an Signature for the Risk of Progression in IgM MGUS to Smoldering/ early precursor stage of the rare lymphoma Waldenström Macroglobulinemia (WM). Although Symptomatic Waldenström comparative gene expression studies on WM, IgM MGUS, and normal B-cells (CTRLs) identified Macroglobulinemia (WM) by a several differentially expressed genes, reliable predictors of progression from IgM MGUS to WM Comparative Transcriptome Analysis have not yet been identified. -

A Deep Transcriptome Meta-Analysis Reveals Sex-Based Molecular Differences in Multiple Sclerosis
medRxiv preprint doi: https://doi.org/10.1101/2021.08.31.21262175; this version posted September 2, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. It is made available under a CC-BY-NC 4.0 International license . A deep transcriptome meta-analysis reveals sex-based molecular differences in Multiple Sclerosis José Francisco Català-Senent1†, Zoraida Andreu2†, Francisco José Roig1,3, Marta R. Hidalgo1, Natalia Yanguas-Casás4, Almudena Neva-Alejo1, Adolfo López-Cerdán5, Irene Soler-Sáez1, María de la Iglesia-Vayá5, Francisco García-García1 Correspondence: Francisco García-García, [email protected] †José Francisco Català-Senent and Zoraida Andreu contributed equally to this study 1Bioinformatics and Biostatistics Unit, Principe Felipe Research Center (CIPF), 46012, Valencia, Spain 2Foundation Valencian Institute of Oncology (FIVO), 46009, Valencia, Spain 3Faculty of Health Sciences. San Jorge University, 50830, Zaragoza, Spain 4Instituto de Investigación Sanitaria Puerta de Hierro-Segovia de Arana (IDIPHISA), Grupo de Investigación en Linfomas, C/Joaquín Rodrigo 2, Majadahonda, 28222 Madrid, Spain 5Biomedical Imaging Unit FISABIO-CIPF, Fundación para el Fomento de la Investigación Sanitaria y Biomédica de la Comunidad Valenciana, 46012, Valencia, Spain NOTE: This preprint reports new research that has not been certified by peer review and should not be used to guide clinical practice. 1 medRxiv preprint doi: https://doi.org/10.1101/2021.08.31.21262175; this version posted September 2, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. -

The Novel Lncrna Lnc-NR2F1 Is Pro- Neurogenic and Mutated in Human
RESEARCH ARTICLE The novel lncRNA lnc-NR2F1 is pro- neurogenic and mutated in human neurodevelopmental disorders Cheen Euong Ang1,2†, Qing Ma3,4,5†, Orly L Wapinski3,4,5†, ShengHua Fan6, Ryan A Flynn3,4,5, Qian Yi Lee1,2, Bradley Coe7, Masahiro Onoguchi3,4,5, Victor Hipolito Olmos1, Brian T Do3, Lynn Dukes-Rimsky6, Jin Xu3, Koji Tanabe1, LiangJiang Wang8, Ulrich Elling9, Josef M Penninger9, Yang Zhao3, Kun Qu3,9§, Evan E Eichler7, Anand Srivastava6,8‡, Marius Wernig1‡*, Howard Y Chang3‡* 1Department of Pathology, Institute for Stem Cell Biology and Regenerative Medicine, Stanford University, Stanford, United States; 2Department of Bioengineering, Stanford University, Stanford, United States; 3Center for Personal Dynamic Regulomes, Stanford University, Stanford, United States; 4Department of Dermatology, Stanford University, Stanford, United States; 5Department of Genetics, Stanford University, Stanford, United States; 6JC Self Research Institute of *For correspondence: Human Genetics, Greenwood Genetic Center, Greenwood, United States; [email protected] (MW); 7Department of Genome Sciences, Howard Hughes Medical Institute, University of [email protected] (HYC) Washington, Seattle, United States; 8Department of Genetics and Biochemistry, †These authors contributed Clemson University, Clemson, United States; 9Institute of Molecular Biotechnology equally to this work of the Austrian Academy of Science (IMBA), Vienna Biocenter, Vienna, Austria ‡These authors also contributed equally to this work Present address: §CAS Key Laboratory -

Introduction to Simvector
LAMP Designer 1.01 Manual PREMIER Biosoft International 3786 Corina Way, Palo Alto, CA 94303-4504 Tel: (650) 856-2703 FAX: (650) 618-1773 E-mail: [email protected] Copyright © 2011 by PREMIER Biosoft International. All Rights Reserved. Information in this manual may change without notice and does not represent a commitment on the part of PREMIER Biosoft International. The software described in this manual is provided by PREMIER Biosoft International under a License Agreement. The software may be used only in accordance with the terms of the agreement. PREMIER Biosoft International ("Premier") claims copyright to this program and documentation as an unpublished work. Claim of copyright does not imply waiver of Premier's other rights. This program and documentation are confidential and the property of Premier. Use, examination, reproduction, copying, decompilation, transfer, and/or disclosure to others are strictly prohibited except by express written agreement with Premier. For more information on PrimerPlex and product updates, visit the PREMIER Biosoft web site at http://www.PremierBiosoft.com. ii LAMP Designer 1.01 INTRODUCTION .................................................................................................. 1 Working Environment.....................................................................................................3 Main Window ...................................................................................................................8 LAMP Primer Search Parameters ................................................................................10