Activity-Dependent Modulation of Neuronal Sodium Channel Expression Joshua Peter Klein Yale University
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Entrez Symbols Name Termid Termdesc 117553 Uba3,Ube1c
Entrez Symbols Name TermID TermDesc 117553 Uba3,Ube1c ubiquitin-like modifier activating enzyme 3 GO:0016881 acid-amino acid ligase activity 299002 G2e3,RGD1310263 G2/M-phase specific E3 ubiquitin ligase GO:0016881 acid-amino acid ligase activity 303614 RGD1310067,Smurf2 SMAD specific E3 ubiquitin protein ligase 2 GO:0016881 acid-amino acid ligase activity 308669 Herc2 hect domain and RLD 2 GO:0016881 acid-amino acid ligase activity 309331 Uhrf2 ubiquitin-like with PHD and ring finger domains 2 GO:0016881 acid-amino acid ligase activity 316395 Hecw2 HECT, C2 and WW domain containing E3 ubiquitin protein ligase 2 GO:0016881 acid-amino acid ligase activity 361866 Hace1 HECT domain and ankyrin repeat containing, E3 ubiquitin protein ligase 1 GO:0016881 acid-amino acid ligase activity 117029 Ccr5,Ckr5,Cmkbr5 chemokine (C-C motif) receptor 5 GO:0003779 actin binding 117538 Waspip,Wip,Wipf1 WAS/WASL interacting protein family, member 1 GO:0003779 actin binding 117557 TM30nm,Tpm3,Tpm5 tropomyosin 3, gamma GO:0003779 actin binding 24779 MGC93554,Slc4a1 solute carrier family 4 (anion exchanger), member 1 GO:0003779 actin binding 24851 Alpha-tm,Tma2,Tmsa,Tpm1 tropomyosin 1, alpha GO:0003779 actin binding 25132 Myo5b,Myr6 myosin Vb GO:0003779 actin binding 25152 Map1a,Mtap1a microtubule-associated protein 1A GO:0003779 actin binding 25230 Add3 adducin 3 (gamma) GO:0003779 actin binding 25386 AQP-2,Aqp2,MGC156502,aquaporin-2aquaporin 2 (collecting duct) GO:0003779 actin binding 25484 MYR5,Myo1e,Myr3 myosin IE GO:0003779 actin binding 25576 14-3-3e1,MGC93547,Ywhah -

Aquaporin Channels in the Heart—Physiology and Pathophysiology
International Journal of Molecular Sciences Review Aquaporin Channels in the Heart—Physiology and Pathophysiology Arie O. Verkerk 1,2,* , Elisabeth M. Lodder 2 and Ronald Wilders 1 1 Department of Medical Biology, Amsterdam University Medical Centers, University of Amsterdam, 1105 AZ Amsterdam, The Netherlands; [email protected] 2 Department of Experimental Cardiology, Amsterdam University Medical Centers, University of Amsterdam, 1105 AZ Amsterdam, The Netherlands; [email protected] * Correspondence: [email protected]; Tel.: +31-20-5664670 Received: 29 March 2019; Accepted: 23 April 2019; Published: 25 April 2019 Abstract: Mammalian aquaporins (AQPs) are transmembrane channels expressed in a large variety of cells and tissues throughout the body. They are known as water channels, but they also facilitate the transport of small solutes, gasses, and monovalent cations. To date, 13 different AQPs, encoded by the genes AQP0–AQP12, have been identified in mammals, which regulate various important biological functions in kidney, brain, lung, digestive system, eye, and skin. Consequently, dysfunction of AQPs is involved in a wide variety of disorders. AQPs are also present in the heart, even with a specific distribution pattern in cardiomyocytes, but whether their presence is essential for proper (electro)physiological cardiac function has not intensively been studied. This review summarizes recent findings and highlights the involvement of AQPs in normal and pathological cardiac function. We conclude that AQPs are at least implicated in proper cardiac water homeostasis and energy balance as well as heart failure and arsenic cardiotoxicity. However, this review also demonstrates that many effects of cardiac AQPs, especially on excitation-contraction coupling processes, are virtually unexplored. -

Hereditary Kidney Disorders
A. Stavljenić-Rukavina Hereditary kidney disorders How to Cite this article: Hereditary Kidney Disorders- eJIFCC 20/01 2009 http://www.ifcc.org 5. HEREDITARY KIDNEY DISORDERS Ana Stavljenić-Rukavina 5.1 Introduction Hereditary kidney disorders represent significant risk for the development of end stage renal desease (ESRD). Most of them are recognized in childhood, or prenataly particularly those phenotypicaly expressed as anomalies on ultrasound examination (US) during pregnancy. They represent almost 50% of all fetal malformations detected by US (1). Furthermore many of urinary tract malformations are associated with renal dysplasia which leeds to renal failure. Recent advances in molecular genetics have made a great impact on better understanding of underlying molecular mechanisms in different kidney and urinary tract disorders found in childhood or adults. Even some of clinical syndromes were not recognized earlier as genetic one. In monogenic kidney diseases gene mutations have been identified for Alport syndrome and thin basement membrane disease, autosomal dominant polycystic kidney disease, and tubular transporter disorders. There is evident progress in studies of polygenic renal disorders as glomerulopathies and diabetic nephropathy. The expanded knowledge on renal physiology and pathophysiology by analyzing the phenotypes caused by defected genes might gain to earlier diagnosis and provide new diagnostic and prognostic tool. The global increasing number of patients with ESRD urges the identification of molecular pathways involved in renal pathophysiology in order to serve as targets for either prevention or intervention. Molecular genetics nowadays possess significant tools that can be used to identify genes involved in renal disease including gene expression arrays, linkage analysis and association studies. -

Variants in the KCNE1 Or KCNE3 Gene and Risk of Ménière’S Disease: a Meta-Analysis
Journal of Vestibular Research 25 (2015) 211–218 211 DOI 10.3233/VES-160569 IOS Press Variants in the KCNE1 or KCNE3 gene and risk of Ménière’s disease: A meta-analysis Yuan-Jun Li, Zhan-Guo Jin and Xian-Rong Xu∗ The Center of Clinical Aviation Medicine, General Hospital of Air Force, Beijing, China Received 1 August 2015 Accepted 8 December 2015 Abstract. BACKGROUND: Ménière’s disease (MD) is defined as an idiopathic disorder of the inner ear characterized by the triad of tinnitus, vertigo, and sensorineural hearing loss. Although many studies have evaluated the association between variants in the KCNE1 or KCNE3 gene and MD risk, debates still exist. OBJECTIVE: Our aim is to evaluate the association between KCNE gene variants, including KCNE1 rs1805127 and KCNE3 rs2270676, and the risk of MD by a systematic review. METHODS: We searched the literature in PubMed, SCOPUS and EMBASE through May 2015. We calculated pooled odds ra- tios (OR) and 95% confidence intervals (CIs) using a fixed-effects model or a random-effects model for the risk to MD associated with different KCNE gene variants. The heterogeneity assumption decided the effect model. RESULTS: A total of three relevant studies, with 302 MD cases and 515 controls, were included in this meta-analysis. The results indicated that neither the KCNE1 rs1805127 variant (for G vs. A: OR = 0.724, 95%CI 0.320, 1.638, P = 0.438), nor the KCNE3 rs2270676 variant (for T vs. C: OR = 0.714, 95%CI 0.327, 1.559, P = 0.398) was associated with MD risk. -

Inherited Renal Tubulopathies—Challenges and Controversies
G C A T T A C G G C A T genes Review Inherited Renal Tubulopathies—Challenges and Controversies Daniela Iancu 1,* and Emma Ashton 2 1 UCL-Centre for Nephrology, Royal Free Campus, University College London, Rowland Hill Street, London NW3 2PF, UK 2 Rare & Inherited Disease Laboratory, London North Genomic Laboratory Hub, Great Ormond Street Hospital for Children National Health Service Foundation Trust, Levels 4-6 Barclay House 37, Queen Square, London WC1N 3BH, UK; [email protected] * Correspondence: [email protected]; Tel.: +44-2381204172; Fax: +44-020-74726476 Received: 11 February 2020; Accepted: 29 February 2020; Published: 5 March 2020 Abstract: Electrolyte homeostasis is maintained by the kidney through a complex transport function mostly performed by specialized proteins distributed along the renal tubules. Pathogenic variants in the genes encoding these proteins impair this function and have consequences on the whole organism. Establishing a genetic diagnosis in patients with renal tubular dysfunction is a challenging task given the genetic and phenotypic heterogeneity, functional characteristics of the genes involved and the number of yet unknown causes. Part of these difficulties can be overcome by gathering large patient cohorts and applying high-throughput sequencing techniques combined with experimental work to prove functional impact. This approach has led to the identification of a number of genes but also generated controversies about proper interpretation of variants. In this article, we will highlight these challenges and controversies. Keywords: inherited tubulopathies; next generation sequencing; genetic heterogeneity; variant classification. 1. Introduction Mutations in genes that encode transporter proteins in the renal tubule alter kidney capacity to maintain homeostasis and cause diseases recognized under the generic name of inherited tubulopathies. -

Molecular Mechanism of Water Channel Aquaporin-2 Trafficking Yumi Noda1,* and Sei Sasaki1
J Pharmacol Sci 96, 249 – 254 (2004) Journal of Pharmacological Sciences ©2004 The Japanese Pharmacological Society Forum Minireview Molecular Mechanisms and Drug Development in Aquaporin Water Channel Diseases: Molecular Mechanism of Water Channel Aquaporin-2 Trafficking Yumi Noda1,* and Sei Sasaki1 1Department of Nephrology, Graduate School, Tokyo Medical and Dental University, Tokyo 113-8519, Japan Received September 29, 2004; Accepted October 13, 2004 Abstract. Targeted positioning of water channel aquaporin-2 (AQP2) strictly regulates body water homeostasis. Trafficking of AQP2 to the apical membrane is critical for the reabsorption of water in renal collecting ducts. Besides the cAMP-mediated effect of vasopressin on AQP2 trafficking to the apical membrane, other signaling cascades also induce this sorting. Recently, AQP2-binding proteins that directly regulate this trafficking have been uncovered: SPA-1, a GTPase-activating protein (GAP) for Rap1, and cytoskeletal protein actin. This review summarizes recent advances related to the trafficking mechanism of AQP2 and its defect causing nephrogenic diabetes insipidus (NDI). Keywords: aquaporin-2, PKA phosphorylation, missorting, Rho, cytoskeleton Introduction receptors located in the hypothalamus are activated and stimulate the secretion of antidiuretic hormone arginine Body water homeostasis is essential for survival of vasopressin (AVP) from the posterior pituitary (22, 23). mammals. Water transport occurs through a specialized After it is released systemically, vasopressin binds to channel called aquaporin (AQP) (1 – 4). AQPs play an vasopressin type 2 receptor (V2R) located on the baso- important role in reabsorption of water and in concentra- lateral membrane of the renal collecting duct principal tion of urine in the kidney. Out of 13 aquaporin iso- cells (24). -

Ubiquitination of Aquaporin-2 in the Kidney
Electrolytes & Blood Pressure 7:1-4, 2009 1 Review article 1) Ubiquitination of Aquaporin-2 in the Kidney Yu-Jung Lee, M.D. and Tae-Hwan Kwon, M.D. Department of Biochemistry and Cell Biology, School of Medicine, Kyungpook National University, Daegu, Korea Ubiquitination is known to be important for endocytosis and lysosomal degradation of aquaporin-2 (AQP2). Ubiquitin (Ub) is covalently attached to the lysine residue of the substrate proteins and activation and attach - ment of Ub to a target protein is mediated by the action of three enzymes (i.e., E1, E2, and E3). In particular, E3 Ub-protein ligases are known to have substrate specificity. This minireview will discuss the ubiquitination of AQP2 and identification of potential E3 Ub-protein ligases for 1-deamino-8-D-arginine vasopressin (dDAVP)-dependent AQP2 regulation. Key Words : kidney tubules, collecting; ubiquitination; vasopressins; aquaporin 2 The kidneys are responsible for the regulation of body This process produces concentrated urine and is essential water and electrolyte metabolism. Thus, understanding of for regulation of body water metabolism 6) . In contrast to the underlying mechanisms for renal water transport is the well-established signaling pathways for the vaso- critical. Water permeability along the nephron has already pressin-regulated AQP2 trafficking and up-regulation of been well characterized in the mammalian kidney 1) . AQP2 expression, the underlying mechanisms for AQP2 Approximately, 180 L/day of glomerular filtrate is gen- endocytosis and intracellular degradation of AQP2 protein erated in an adult human, more than 80-90% of the glomer- are unclear. So far, two hormones (prostaglandin E2 and ular filtrate is constitutively reabsorbed by the highly water dopamine) cause AQP2 internalization independent of permeable proximal tubules and descending thin limbs of S256 dephosphorylation 7, 8) . -

Renal Aquaporins
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Kidney International, Vol. 49 (1996), pp.1712—1717 Renal aquaporins MARK A. KNEPPER, JAMES B. WADE, JAMES TERRIS, CAROLYN A. ECELBARGER, DAVID MARPLES, BEATRICE MANDON, CHUNG-LIN CHOU, B.K. KISHORE, and SØREN NIELSEN Laborato,y of Kidney and Electrolyte Metabolism, National Heart, Lung and Blood Institute, National Institutes of Health, Bethesda, Matyland, USA; Department of Cell Biology, Institute of Anatomy, University of Aarhus, Aarhus, Denmark; and Department of Physiology, University of Maiyland College of Medicine, Baltimore, and Department of Physiology, Unifornied Services University of the Health Sciences, Bethesda, Maiyland, USA Renal aquaporins. Aquaporins (AQPs) are a newly recognized family of gate the localization and regulation of the four renal aquaporins transmembrane proteins that function as molecular water channels. At (AQP1, AQP2, AQP3 and AQP4). least four aquaporins are expressed in the kidney where they mediate Urine is concentrated as a result of the combined function of rapid water transport across water-permeable epithelia and play critical roles in urinary concentrating and diluting processes. AQP1 is constitu- the loop of Henle, which generates a high osmolality in the renal tively expressed at extremely high levels in the proximal tubule and medulla by countercurrent multiplication, and the collecting duct, descending limb of Henle's loop. AQP2, -3 and -4 are expressed predom- which, in the presence of the antidiuretic hormone vasopressin, inantly in the collecting duct system. AQP2 is the predominant water permits osmotic equilibration between the urine and the hyper- channel in the apical plasma membrane and AQP3 and -4arefound in the basolateral plasma membrane. -

AQP3 and AQP5—Potential Regulators of Redox Status in Breast Cancer
molecules Review AQP3 and AQP5—Potential Regulators of Redox Status in Breast Cancer Lidija Milkovi´c and Ana Cipakˇ Gašparovi´c* Division of Molecular Medicine, Ruder¯ Boškovi´cInstitute, HR-10000 Zagreb, Croatia; [email protected] * Correspondence: [email protected]; Tel.: +385-1-457-1212 Abstract: Breast cancer is still one of the leading causes of mortality in the female population. Despite the campaigns for early detection, the improvement in procedures and treatment, drastic improvement in survival rate is omitted. Discovery of aquaporins, at first described as cellular plumbing system, opened new insights in processes which contribute to cancer cell motility and proliferation. As we discover new pathways activated by aquaporins, the more we realize the complexity of biological processes and the necessity to fully understand the pathways affected by specific aquaporin in order to gain the desired outcome–remission of the disease. Among the 13 human aquaporins, AQP3 and AQP5 were shown to be significantly upregulated in breast cancer indicating their role in the development of this malignancy. Therefore, these two aquaporins will be discussed for their involvement in breast cancer development, regulation of oxidative stress and redox signalling pathways leading to possibly targeting them for new therapies. Keywords: AQP3; AQP5; oxidative stress Citation: Milkovi´c,L.; Cipakˇ 1. Introduction Gašparovi´c,A. AQP3 and Despite the progress in research and treatment procedures, cancer still remains the AQP5—Potential Regulators of Redox leading cause of death. Today, cancer is targeted via different approaches which is deter- Status in Breast Cancer. Molecules mined by diagnosis, tumour marker expression, and specific mutations. -

Pflugers Final
CORE Metadata, citation and similar papers at core.ac.uk Provided by Serveur académique lausannois A comprehensive analysis of gene expression profiles in distal parts of the mouse renal tubule. Sylvain Pradervand2, Annie Mercier Zuber1, Gabriel Centeno1, Olivier Bonny1,3,4 and Dmitri Firsov1,4 1 - Department of Pharmacology and Toxicology, University of Lausanne, 1005 Lausanne, Switzerland 2 - DNA Array Facility, University of Lausanne, 1015 Lausanne, Switzerland 3 - Service of Nephrology, Lausanne University Hospital, 1005 Lausanne, Switzerland 4 – these two authors have equally contributed to the study to whom correspondence should be addressed: Dmitri FIRSOV Department of Pharmacology and Toxicology, University of Lausanne, 27 rue du Bugnon, 1005 Lausanne, Switzerland Phone: ++ 41-216925406 Fax: ++ 41-216925355 e-mail: [email protected] and Olivier BONNY Department of Pharmacology and Toxicology, University of Lausanne, 27 rue du Bugnon, 1005 Lausanne, Switzerland Phone: ++ 41-216925417 Fax: ++ 41-216925355 e-mail: [email protected] 1 Abstract The distal parts of the renal tubule play a critical role in maintaining homeostasis of extracellular fluids. In this review, we present an in-depth analysis of microarray-based gene expression profiles available for microdissected mouse distal nephron segments, i.e., the distal convoluted tubule (DCT) and the connecting tubule (CNT), and for the cortical portion of the collecting duct (CCD) (Zuber et al., 2009). Classification of expressed transcripts in 14 major functional gene categories demonstrated that all principal proteins involved in maintaining of salt and water balance are represented by highly abundant transcripts. However, a significant number of transcripts belonging, for instance, to categories of G protein-coupled receptors (GPCR) or serine-threonine kinases exhibit high expression levels but remain unassigned to a specific renal function. -
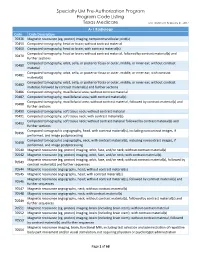
Specialty UM Pre-Authorization Program Code Listing
Specialty UM Pre-Authorization Program Program Code Listing Texas Medicare Last Updated: February 21, 2017 A-1 Radiology Code Code Description 70336 Magnetic resonance (eg, proton) imaging, temporomandibular joint(s) 70450 Computed tomography, head or brain; without contrast material 70460 Computed tomography, head or brain; with contrast material(s) Computed tomography, head or brain; without contrast material, followed by contrast material(s) and 70470 further sections Computed tomography, orbit, sella, or posterior fossa or outer, middle, or inner ear; without contrast 70480 material Computed tomography, orbit, sella, or posterior fossa or outer, middle, or inner ear; with contrast 70481 material(s) Computed tomography, orbit, sella, or posterior fossa or outer, middle, or inner ear; without contrast 70482 material, followed by contrast material(s) and further sections 70486 Computed tomography, maxillofacial area; without contrast material 70487 Computed tomography, maxillofacial area; with contrast material(s) Computed tomography, maxillofacial area; without contrast material, followed by contrast material(s) and 70488 further sections 70490 Computed tomography, soft tissue neck; without contrast material 70491 Computed tomography, soft tissue neck; with contrast material(s) Computed tomography, soft tissue neck; without contrast material followed by contrast material(s) and 70492 further sections Computed tomographic angiography, head, with contrast material(s), including noncontrast images, if 70496 performed, and image postprocessing -

Mechanisms and Regulation of Cardiac Cav1.2 Trafficking
International Journal of Molecular Sciences Review Mechanisms and Regulation of Cardiac CaV1.2 Trafficking Maartje Westhoff and Rose E. Dixon * Department of Physiology and Membrane Biology, School of Medicine, University of California, Davis, CA 95616, USA; [email protected] * Correspondence: [email protected] Abstract: During cardiac excitation contraction coupling, the arrival of an action potential at the 2+ ventricular myocardium triggers voltage-dependent L-type Ca (CaV1.2) channels in individual myocytes to open briefly. The level of this Ca2+ influx tunes the amplitude of Ca2+-induced Ca2+ release from ryanodine receptors (RyR2) on the junctional sarcoplasmic reticulum and thus the magnitude of the elevation in intracellular Ca2+ concentration and ultimately the downstream contraction. The number and activity of functional CaV1.2 channels at the t-tubule dyads dictates the amplitude of the Ca2+ influx. Trafficking of these channels and their auxiliary subunits to the cell surface is thus tightly controlled and regulated to ensure adequate sarcolemmal expression to sustain this critical process. To that end, recent discoveries have revealed the existence of internal reservoirs of preformed CaV1.2 channels that can be rapidly mobilized to enhance sarcolemmal expression in times of acute stress when hemodynamic and metabolic demand increases. In this review, we provide an overview of the current thinking on CaV1.2 channel trafficking dynamics in the heart. We highlight the numerous points of control including the biosynthetic pathway, the endosomal recycling pathway, ubiquitination, and lysosomal and proteasomal degradation pathways, and discuss the effects of β-adrenergic and angiotensin receptor signaling cascades on this process. Keywords: L-type calcium channels; ion channel trafficking; t-tubule; caveolae; calcium signaling; Citation: Westhoff, M.; Dixon, R.E.