Pathway Tools Version 24.0: Integrated Software for Pathway/Genome Informatics and Systems Biology
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Functional Aspects and Genomic Analysis
Discussion IV./ Discussion IV.1. The discovery of UEV protein and its role in different cellular processes In our studies of cell differentiation and cell cycle control, we have isolated a new gene that is downregulated upon cell differentiation. We have demonstrated that this gene, previously called CROC1 and considered a transcriptional activator of c-fos (Rothofsky and Lin, 1997), is highly conserved in phylogeny, and constitutes, by sequence relationship, a novel subfamily of the ubiquitin-conjugating, or E2, enzymes. We have demonstrated that these proteins are very conserved in all eukaryotic organisms and that they are very similar to the E2 enzymes in sequence and structure, but these proteins lack a conserved cysteine residue responsible for the catalytic activity of the E2 enzymes (Chen et al., 1993; Jentsch, 1992a). We have given these proteins the name UEV (ubiquitin-conjugating E2 enzyme variant). Work by other laboratories has shown that experimental mutagenesis of this cysteine in the catalytic center leads to the inactivation of the E2 enzyme activity, and the mutated protein can behave as a dominant negative variant (Banerjee et al., 1995; Sung et al., 1990; Madura et al., 1993). However, in our initial experiments with recombinant proteins, UEV did not behave as a negative regulator of ubiquitination (Sancho et al., 1998). We have demonstrated also the existence of at least two different human UEV genes, one coding for UEV1/CROC-1, and the other coding for UEV2. The second protein has also been given different names by others, DDVit-1 (Fritsche et al., 1997). 30 Discussion The transcripts from the two human UEV genes differ in their 3’untranslated regions, and produce almost identical proteins. -

Pathways and Networks Biological Meaning of the Gene Sets
Pathways and Networks Biological meaning of the gene sets Gene ontology terms ? Pathway mapping Linking to Pubmed abstracts or associted MESH terms Regulation by the same transcription factor (module) Protein families and domains Gene set enrichment analysis Over representation analysis 1 Gene set enrichment analysis 1. Given an a priori defined set of genes S. 2. Rank genes (e.g. by t‐value between 2 groups of microarray samples) ranked gene list L. 3. Calculation of an enrichment score (ES) that reflects the degree to which a set S is overrepresented at the extremes (top or bottom) of the entire ranked list L. 4. Estimation the statistical significance (nominal P value) of the ES by using an empirical phenotype‐based permutation test procedure. 5. Adjustment for multiple hypothesis testing by controlling the false discovery rate (FDR). Gene set enrichment analysis Subramanian A et al. Proc Natl Acad Sci (2005) 2 Biochemical and Metabolic Pathways Böhringer Mannheim Signaling networks (in cancer cell) Hannah, Weinberg, Cell. 2000 137 NCI curated pathway maps (http://pid.nci.nih.gov) Signal Transduction Knowledge Environment http://stke.sciencemag.org/cm/ 3 Pathways • Pathways are available mechanistic information •Kyoto Encylopedia of Genes and Genomes, KEGG (http://www.genome.jp/kegg/) BioCYc • EcoCYC Metabolic Pathway Database (E. Coli) •Also for other organisms (e.g. HumanCYC) 4 Pathways • Pathways from Biocarta • http://www.biocarta.com/genes/index.asp Transpath Part of larger BioBase package (commercial) • PathwayBuilder package -

University of Maryland Establishes Center for Health Related Informatics and Bioimaging
University of Maryland Establishes Center for Health Related Informatics and Bioimaging News & Events UNIVERSITY OF MARYLAND ESTABLISHES CENTER FOR HEALTH CONTACT RELATED INFORMATICS AND BIOIMAGING Monday, June 10, 2013 Karen Robinson, Media Relations New Center, Led By Distinguished Bioinformatics Scientist Owen White, (410) 706-7590 Ph.D., Unites Scientists Across Disciplines to Integrate Genomics and Personalized Medicine into Research and Clinical Care [email protected] University of Maryland, Baltimore campus President Jay A. Perman, M.D., and Media Relations Team University of Maryland School of Medicine Dean E. Albert Reece, M.D., Ph.D., M.B.A., wish to announce the establishment of a new center to unite research scientists and physicians across disciplines. The center will employ these SEARCH ARTICLES interdisciplinary connections to enhance the use of cutting edge medical science such as genomics and personalized medicine to accelerate research discoveries and improve health care outcomes. Participants in the new News Archives University of Maryland Center for Health-Related Informatics and Bioimaging (CHIB) will collaborate with computer scientists, engineers, life scientists and others at a similar center at the University of Maryland, College Park campus, CHIB Co-Director Owen White, Ph.D. together forming a joint center supported by the M-Power Maryland initiative. LEARN MORE University of Maryland School of Medicine Dean E. Albert Reece, M.D., Ph.D., M.B.A., with the concurrence of President Perman, has appointed as co-director of the new center Owen White, Ph.D., Professor of Epidemiology and Public Health and Director of Bioinformatics at the University of Maryland School of Medicine Institute for Genome Sciences. -

Computational Analysis and Predictive Cheminformatics Modeling of Small Molecule Inhibitors of Epigenetic Modifiers
RESEARCH ARTICLE Computational Analysis and Predictive Cheminformatics Modeling of Small Molecule Inhibitors of Epigenetic Modifiers Salma Jamal1☯‡, Sonam Arora2☯‡, Vinod Scaria3* 1 CSIR Open Source Drug Discovery Unit (CSIR-OSDD), Anusandhan Bhawan, Delhi, India, 2 Delhi Technological University, Delhi, India, 3 GN Ramachandran Knowledge Center for Genome Informatics, CSIR Institute of Genomics and Integrative Biology (CSIR-IGIB), Delhi, India ☯ These authors contributed equally to this work. ‡ These authors are joint first authors on this work. * [email protected] a11111 Abstract Background The dynamic and differential regulation and expression of genes is majorly governed by the OPEN ACCESS complex interactions of a subset of biomolecules in the cell operating at multiple levels start- Citation: Jamal S, Arora S, Scaria V (2016) ing from genome organisation to protein post-translational regulation. The regulatory layer Computational Analysis and Predictive contributed by the epigenetic layer has been one of the favourite areas of interest recently. Cheminformatics Modeling of Small Molecule Inhibitors of Epigenetic Modifiers. PLoS ONE 11(9): This layer of regulation as we know today largely comprises of DNA modifications, histone e0083032. doi:10.1371/journal.pone.0083032 modifications and noncoding RNA regulation and the interplay between each of these major Editor: Wei Yan, University of Nevada School of components. Epigenetic regulation has been recently shown to be central to development Medicine, UNITED STATES of a number of disease processes. The availability of datasets of high-throughput screens Received: June 10, 2013 for molecules for biological properties offer a new opportunity to develop computational methodologies which would enable in-silico screening of large molecular libraries. -

Unicarbkb: Building a Standardised and Scalable Informatics Platform for Glycosciences Research
Platforms EOI: UniCarbKB: Building a Standardised and Scalable Informatics Platform for Glycosciences Research 20 September 2019 at 13:02 Project title UniCarbKB: Building a Standardised and Scalable Informatics Platform for Glycosciences Research Field of Research code(s) 03 CHEMICAL SCIENCES 06 BIOLOGICAL SCIENCES 08 INFORMATION AND COMPUTING SCIENCES 11 MEDICAL AND HEALTH SCIENCES EOI Lead Name Matthew Campbell EOI lead Research Group Institute for Glycomics EOI lead Organisation Institute for Glycomics, Griffith University EOI lead Email Collaborator details Name Research Group Organisation Malcolm Wolski eResearch Services Griffith University Project description Over the last few years the glycomics knowledge platform UniCarbKB has contributed to the growth of international glycoinformatics resources by providing access to data collections, web services and software applications supported by biocuration activities. However, UniCarbKB has continued to develop as a single monolithic resource. This proposed project will improve the growth and scalability of UniCarbKB by breaking down the platform into smaller and composable pieces. This will be achieved through the adoption of a more decentralised approach and a microservice architecture that will allow components of the platform to scale at different rates. This architecture will allow us is to build an extendable and cross-disciplinary resource by integrating existing data with new analysis tools. The adoption of international resources (e.g. NIH GlyGen), will allow not just mapping of glycan data to genes and proteins but also pathways, gene ontology, publications and a wide variety of data from EBI, NCBI and other sources. Additionally, we propose to integrate glycan array data and develop ontologies that can serve as tools for integration and subsequently analysis of glycan-associated Existing technology Adopt The proposed project will adopt a multi-tier application architecture in which applications will be developed and deployed as microservices. -

Corynebactin and Enterobactin: Related Siderophores of Opposite Chirality Martin E
Published on Web 02/20/2002 Corynebactin and Enterobactin: Related Siderophores of Opposite Chirality Martin E. Bluhm, Sanggoo S. Kim, Emily A. Dertz, and Kenneth N. Raymond* Department of Chemistry, UniVersity of California, Berkeley, California 94720-1460 Received July 18, 2001 The major role of siderophores in microbial iron transport and bacterial pathogenicity is now well established.1 These low mo- lecular weight chelators are expressed to overcome the insolubility of Fe3+ at pH 7 (∼10-18 M).2 The ferric complexes enter the bacterial cell via specific receptor proteins on the cell membrane. Once incorporated, iron is released via reduction, hydrolysis, or ligand-exchange mechanisms.3 Enterobactin (1) is produced by both Gram-negative4a,b and Gram-positive4c bacteria, and has an ex- 49 5 traordinarily high stability (Kf ) 10 ) with metal coordination at neutral pH accomplished through the six catecholate oxygens.6 The Figure 1. Siderophores enterobactin (1) and corynebactin (2). The synthetic chirality of the iron center in enterobactin is ∆,6 and this chirality, analogue 7 is a hybrid, composed of the serine trilactone connected to the while not essential for receptor recognition and outer membrane side chain of corynebactin. The triserine trilactone trihydrochloride (5) was used for the preparation of 7. transport,7 is essential for iron utilization; the mirror image enantio- enterobactin complex does not promote microbial growth.8 Recently Scheme 1. Synthesis of serine corynebactin (7) a closely related siderophore, corynebactin (2), was found to be produced by the Gram-positive Corynebacterium glutamicum.9 Both siderophores are based on a trilactone backbone, consisting of L-serine units in enterobactin and L-threonine units in corynebactin. -
Hierarchical Classification of Gene Ontology Terms Using the Gostruct
Hierarchical classification of Gene Ontology terms using the GOstruct method Artem Sokolov and Asa Ben-Hur Department of Computer Science, Colorado State University Fort Collins, CO, 80523, USA Abstract Protein function prediction is an active area of research in bioinformatics. And yet, transfer of annotation on the basis of sequence or structural similarity remains widely used as an annotation method. Most of today's machine learning approaches reduce the problem to a collection of binary classification problems: whether a protein performs a particular function, sometimes with a post-processing step to combine the binary outputs. We propose a method that directly predicts a full functional annotation of a protein by modeling the structure of the Gene Ontology hierarchy in the framework of kernel methods for structured-output spaces. Our empirical results show improved performance over a BLAST nearest-neighbor method, and over algorithms that employ a collection of binary classifiers as measured on the Mousefunc benchmark dataset. 1 Introduction Protein function prediction is an active area of research in bioinformatics [25]; and yet, transfer of annotation on the basis of sequence or structural similarity remains the standard way of assigning function to proteins in newly sequenced organisms [20]. The Gene Ontology (GO), which is the current standard for annotating gene products and proteins, provides a large set of terms arranged in a hierarchical fashion that specify a gene-product's molecular function, the biological process it is involved in, and its localization to a cellular component [12]. GO term prediction is therefore a hierarchical classification problem, made more challenging by the thousands of annotation terms available. -

Bioinformatics Study of Lectins: New Classification and Prediction In
Bioinformatics study of lectins : new classification and prediction in genomes François Bonnardel To cite this version: François Bonnardel. Bioinformatics study of lectins : new classification and prediction in genomes. Structural Biology [q-bio.BM]. Université Grenoble Alpes [2020-..]; Université de Genève, 2021. En- glish. NNT : 2021GRALV010. tel-03331649 HAL Id: tel-03331649 https://tel.archives-ouvertes.fr/tel-03331649 Submitted on 2 Sep 2021 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. THÈSE Pour obtenir le grade de DOCTEUR DE L’UNIVERSITE GRENOBLE ALPES préparée dans le cadre d’une cotutelle entre la Communauté Université Grenoble Alpes et l’Université de Genève Spécialités: Chimie Biologie Arrêté ministériel : le 6 janvier 2005 – 25 mai 2016 Présentée par François Bonnardel Thèse dirigée par la Dr. Anne Imberty codirigée par la Dr/Prof. Frédérique Lisacek préparée au sein du laboratoire CERMAV, CNRS et du Computer Science Department, UNIGE et de l’équipe PIG, SIB Dans les Écoles Doctorales EDCSV et UNIGE Etude bioinformatique des lectines: nouvelle classification et prédiction dans les génomes Thèse soutenue publiquement le 8 Février 2021, devant le jury composé de : Dr. Alexandre de Brevern UMR S1134, Inserm, Université Paris Diderot, Paris, France, Rapporteur Dr. -
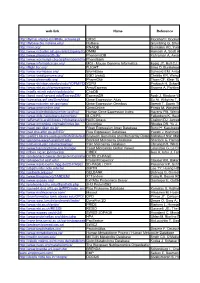
Web-Link Name Reference DRSC Flockhart I, Booker M, Kiger A, Et Al.: Flyrnai: the Drosophila Rnai Screening Center Database
web-link Name Reference http://flyrnai.org/cgi-bin/RNAi_screens.pl DRSC Flockhart I, Booker M, Kiger A, et al.: FlyRNAi: the Drosophila RNAi screening center database. Nucleic Acids Res. 34(Database issue):D489-94 (2006) http://flybase.bio.indiana.edu/ FlyBase Grumbling G, Strelets V.: FlyBase: anatomical data, images and queries. Nucleic Acids Res. 34(Database issue):D484-8 (2006) http://rnai.org/ RNAiDB Gunsalus KC, Yueh WC, MacMenamin P, et al.: RNAiDB and PhenoBlast: web tools for genome-wide phenotypic mapping projects. Nucleic Acids Res. 32(Database issue):D406-10 (2004) http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=OMIMOMIM Hamosh A, Scott AF, Amberger JS, et al.: Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 33(Database issue):D514-7 (2005) http://www.phenomicdb.de/ PhenomicDB Kahraman A, Avramov A, Nashev LG, et al.: PhenomicDB: a multi-species genotype/phenotype database for comparative phenomics. Bioinformatics 21(3):418-20 (2005) http://www.worm.mpi-cbg.de/phenobank2/cgi-bin/MenuPage.pyPhenoBank http://www.informatics.jax.org/ MGI - Mouse Genome Informatics Eppig JT, Bult CJ, Kadin JA, et al.: The Mouse Genome Database (MGD): from genes to mice--a community resource for mouse biology. Nucleic Acids Res. 33(Database issue):D471-5 (2005) http://flight.licr.org/ FLIGHT Sims D, Bursteinas B, Gao Q, et al.: FLIGHT: database and tools for the integration and cross-correlation of large-scale RNAi phenotypic datasets. Nucleic Acids Res. 34(Database issue):D479-83 (2006) http://www.wormbase.org/ WormBase Schwarz EM, Antoshechkin I, Bastiani C, et al.: WormBase: better software, richer content. -

Toolboxes for a Standardised and Systematic Study of Glycans
Campbell et al. BMC Bioinformatics 2014, 15(Suppl 1):S9 http://www.biomedcentral.com/1471-2105/15/S1/S9 RESEARCH Open Access Toolboxes for a standardised and systematic study of glycans Matthew P Campbell1, René Ranzinger2, Thomas Lütteke3, Julien Mariethoz4, Catherine A Hayes5, Jingyu Zhang1, Yukie Akune6, Kiyoko F Aoki-Kinoshita6, David Damerell7,11, Giorgio Carta8, Will S York2, Stuart M Haslam7, Hisashi Narimatsu9, Pauline M Rudd8, Niclas G Karlsson4, Nicolle H Packer1, Frédérique Lisacek4,10* From Integrated Bio-Search: 12th International Workshop on Network Tools and Applications in Biology (NETTAB 2012) Como, Italy. 14-16 November 2012 Abstract Background: Recent progress in method development for characterising the branched structures of complex carbohydrates has now enabled higher throughput technology. Automation of structure analysis then calls for software development since adding meaning to large data collections in reasonable time requires corresponding bioinformatics methods and tools. Current glycobioinformatics resources do cover information on the structure and function of glycans, their interaction with proteins or their enzymatic synthesis. However, this information is partial, scattered and often difficult to find to for non-glycobiologists. Methods: Following our diagnosis of the causes of the slow development of glycobioinformatics, we review the “objective” difficulties encountered in defining adequate formats for representing complex entities and developing efficient analysis software. Results: Various solutions already implemented and strategies defined to bridge glycobiology with different fields and integrate the heterogeneous glyco-related information are presented. Conclusions: Despite the initial stage of our integrative efforts, this paper highlights the rapid expansion of glycomics, the validity of existing resources and the bright future of glycobioinformatics. -

Maternally Inherited Pirnas Direct Transient Heterochromatin Formation
RESEARCH ARTICLE Maternally inherited piRNAs direct transient heterochromatin formation at active transposons during early Drosophila embryogenesis Martin H Fabry, Federica A Falconio, Fadwa Joud, Emily K Lythgoe, Benjamin Czech*, Gregory J Hannon* CRUK Cambridge Institute, University of Cambridge, Li Ka Shing Centre, Cambridge, United Kingdom Abstract The PIWI-interacting RNA (piRNA) pathway controls transposon expression in animal germ cells, thereby ensuring genome stability over generations. In Drosophila, piRNAs are intergenerationally inherited through the maternal lineage, and this has demonstrated importance in the specification of piRNA source loci and in silencing of I- and P-elements in the germ cells of daughters. Maternally inherited Piwi protein enters somatic nuclei in early embryos prior to zygotic genome activation and persists therein for roughly half of the time required to complete embryonic development. To investigate the role of the piRNA pathway in the embryonic soma, we created a conditionally unstable Piwi protein. This enabled maternally deposited Piwi to be cleared from newly laid embryos within 30 min and well ahead of the activation of zygotic transcription. Examination of RNA and protein profiles over time, and correlation with patterns of H3K9me3 deposition, suggests a role for maternally deposited Piwi in attenuating zygotic transposon expression in somatic cells of the developing embryo. In particular, robust deposition of piRNAs targeting roo, an element whose expression is mainly restricted to embryonic development, results in the deposition of transient heterochromatic marks at active roo insertions. We hypothesize that *For correspondence: roo, an extremely successful mobile element, may have adopted a lifestyle of expression in the [email protected] embryonic soma to evade silencing in germ cells. -

To Find Information About Arabidopsis Genes Leonore Reiser1, Shabari
UNIT 1.11 Using The Arabidopsis Information Resource (TAIR) to Find Information About Arabidopsis Genes Leonore Reiser1, Shabari Subramaniam1, Donghui Li1, and Eva Huala1 1Phoenix Bioinformatics, Redwood City, CA USA ABSTRACT The Arabidopsis Information Resource (TAIR; http://arabidopsis.org) is a comprehensive Web resource of Arabidopsis biology for plant scientists. TAIR curates and integrates information about genes, proteins, gene function, orthologs gene expression, mutant phenotypes, biological materials such as clones and seed stocks, genetic markers, genetic and physical maps, genome organization, images of mutant plants, protein sub-cellular localizations, publications, and the research community. The various data types are extensively interconnected and can be accessed through a variety of Web-based search and display tools. This unit primarily focuses on some basic methods for searching, browsing, visualizing, and analyzing information about Arabidopsis genes and genome, Additionally we describe how members of the community can share data using TAIR’s Online Annotation Submission Tool (TOAST), in order to make their published research more accessible and visible. Keywords: Arabidopsis ● databases ● bioinformatics ● data mining ● genomics INTRODUCTION The Arabidopsis Information Resource (TAIR; http://arabidopsis.org) is a comprehensive Web resource for the biology of Arabidopsis thaliana (Huala et al., 2001; Garcia-Hernandez et al., 2002; Rhee et al., 2003; Weems et al., 2004; Swarbreck et al., 2008, Lamesch, et al., 2010, Berardini et al., 2016). The TAIR database contains information about genes, proteins, gene expression, mutant phenotypes, germplasms, clones, genetic markers, genetic and physical maps, genome organization, publications, and the research community. In addition, seed and DNA stocks from the Arabidopsis Biological Resource Center (ABRC; Scholl et al., 2003) are integrated with genomic data, and can be ordered through TAIR.