An Aromatic Acetamide Derivative System As an Example
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Overview of VOC Emissions and Chemistry from PTR
Open Access Atmos. Chem. Phys. Discuss., 15, C7547–C7558, 2015 Atmospheric www.atmos-chem-phys-discuss.net/15/C7547/2015/ Chemistry © Author(s) 2015. This work is distributed under the Creative Commons Attribute 3.0 License. and Physics Discussions Interactive comment on “Overview of VOC emissions and chemistry from PTR-TOF-MS measurements during the SusKat-ABC campaign: high acetaldehyde, isoprene and isocyanic acid in wintertime air of the Kathmandu Valley” by C. Sarkar et al. Anonymous Referee #1 Received and published: 3 October 2015 The study produced a lot of important, unique content of great interest to ACP read- ers. However, the presentation is not quite there. For example, the references have unwanted numbers appended throughout. More importantly, the length should be cut in half while both clarifying and focusing on the main points that are most strongly sup- ported as discussed below. Some other summary thoughts about authors presumed key points in conclusions and abstract: C7547 1. First deployment of PTR-TOF-MS in South Asia, This is very significant as are the high levels measured of certain species noted below. 2. 71 ion peaks detected, of which 37 had campaign average concentrations greater than 200 ppt, which highlights chemical complexity of the Kathmandu Valley‘s air. Great, but briefly, why the 200 ppt cut-off? 3. Acetaldehyde, acetonitrile, isoprene concentrations were among the highest recorded in the world. They are among the highest reported for urban ambi- ent air. Urban area isoprene could be overestimated due to isomers from other sources. For instance, in smoke the “isoprene peak” is 20 % penetenes. -

Research and Mechanism of Two-Step Preparation of Acetamide from Microalgae Under Hydrothermal Conditions
E3S Web of Conferences 194, 02006 (2020) https://doi.org/10.1051/e3sconf/202019402006 ICAEER 2020 Research and Mechanism of Two-step Preparation of Acetamide from Microalgae under Hydrothermal Conditions Jikang Jiang1, Haixia Ye2, Jingfei Zhou1, Chenlu Wang1, Zheng Shen1, 3, *, and Yalei Zhang1, 3 1National Engineering Research Center of Protected Agriculture, Tongji University, Shanghai 200092, China 2Shanghai Huajie Ecological Environment Engineering Co., Ltd., Shanghai, 201407, China 3State Key Laboratory of Pollution Control and Resource Reuse, Tongji University, Shanghai 200092, China Abstract. A two-step synthesis of acetamide under hydrothermal condition from microalgae, is presented. results showed that the best yield of acetamide and selectivity of acetic acid were 9.5 % and 60.1 % at 320 ℃ for 8 min with a NH3 supply of 30. Algae such as spirulina, cyanobacteria and autotrophic chlorella could also acquire acetamide, and lactic acid was found to be an important intermediate during the exploration of reaction pathways. These results demonstrated that it is possible to develop a process for conversion of microalgae biomass into acetamide. 1 Introduction carbohydrates [17-19]. Most importantly, compared to other ways, the hydrothermal treatment of microalgae Diminishing fossil fuel reserves and ever-increasing CO2 does not require dehydration, which could save a great (carbon dioxide) concentration have prompted the deal of energy in the period of pretreatment [1, 19]. exploration for an alternate renewable and sustainable Acetic acid is one of main and stable intermediates energy source [1]. Meanwhile, the treatment of algae mud during hydrothermal oxidation of organics [1], which caused by algae bloom has been the focus of research in contributes it to be the target product for organic waste recent years [1]. -

Safety Data Sheet According to 29CFR1910/1200 and GHS Rev
Safety Data Sheet according to 29CFR1910/1200 and GHS Rev. 3 Effective date : 10.24.2014 Page 1 of 7 Acetamide Crystal, Tech Grade SECTION 1 : Identification of the substance/mixture and of the supplier Product name : Acetamide Crystal, Tech Grade Manufacturer/Supplier Trade name: Manufacturer/Supplier Article number: S25116 Recommended uses of the product and uses restrictions on use: Manufacturer Details: AquaPhoenix Scientific 9 Barnhart Drive, Hanover, PA 17331 Supplier Details: Fisher Science Education 15 Jet View Drive, Rochester, NY 14624 Emergency telephone number: Fisher Science Education Emergency Telephone No.: 800-535-5053 SECTION 2 : Hazards identification Classification of the substance or mixture: Health hazard Carcinogenicity, category 2 Hazards Not Otherwise Classified - Combustible Dust Carcinogenicity - Category 2 Signal word :Warning Hazard statements: Suspected of causing cancer Precautionary statements: If medical advice is needed, have product container or label at hand Keep out of reach of children Read label before use Do not eat, drink or smoke when using this product Obtain special instructions before use Do not handle until all safety precautions have been read and understood Use personal protective equipment as required IF exposed or concerned: Get medical advice/attention Store locked up Dispose of contents and container as instructed in Section 13 Combustible Dust Hazard: : May form combustible dust concentrations in air (during processing). Other Non-GHS Classification: WHMIS Created by Global Safety Management, -

Conversion of Indole-3-Acetaldehyde to Indole-3-Acetic Acid in Cell-Wall Fraction of Barley {Hordeum Vulgare) Seedlings
Plant Cell Physiol. 38(3): 268-273 (1997) JSPP © 1997 Conversion of Indole-3-Acetaldehyde to Indole-3-Acetic Acid in Cell-Wall Fraction of Barley {Hordeum vulgare) Seedlings Ken-ichi Tsurusaki1, Kazuyoshi Takeda2 and Naoki Sakurai3 1 Faculty of Liberal Arts, Fukuyama University, Fukuyama, 729-02 Japan 2 Research Institute for Bioresources, Okayama University, Kurashiki, Okayama, 710 Japan 3 Department of Environmental Studies, Faculty of Integrated Arts & Sciences, Hiroshima University, Higashi-Hiroshima, 739 Japan The cell-wall fraction of barley seedlings was able (Trp) has been suggested as a primary precursor of IAA to oxidize indole-3-acetaldehyde (IAAld) to form IAA, (Gordon 1954, Gibson et al. 1972, Monteiro et al. 1988, whereas the fraction did not catalyze the conversion of in- Cooney and Nonhebel 1991, Bialek et al. 1992, Koshiba dole-3-acetonitrile or indole-3-acetamide to IAA. The activ- and Matsuyama 1993, Koshiba et al. 1995), because Trp iDownloaded from https://academic.oup.com/pcp/article/38/3/268/1928462 by guest on 24 September 2021 s ity was lower in a semi-dwarf mutant that had an endog- similar in structure to IAA and is ubiquitous in plant enous IAA level lower than that of the normal isogenic tissues. strain [Inouhe et al. (1982) Plant Cell Physiol. 23: 689]. Two pathways of IAA biosynthesis from L-Trp have The soluble fraction also contained some activity; the activ- been proposed in higher plants: Trp —• indole-3-pyruvic ity was similar in the normal and mutant strains. The op- acid -»indole-3-acetaldehyde (IAAld) ->• IAA; or Trp -> timal pH for the conversion of IAAld to IAA in the cell- tryptamine —• IAAld -* IAA. -

Preparation and Characterization of the Enol of Acetamide: 1-Aminoethenol, a High-Energy Cite This: Chem
Chemical Science View Article Online EDGE ARTICLE View Journal | View Issue Preparation and characterization of the enol of acetamide: 1-aminoethenol, a high-energy Cite this: Chem. Sci., 2020, 11,12358 † All publication charges for this article prebiotic molecule have been paid for by the Royal Society of Chemistry Artur Mardyukov, Felix Keul and Peter R. Schreiner * Amide tautomers, which constitute the higher-energy amide bond linkage, not only are key for a variety of biological but also prebiotic processes. In this work, we present the gas-phase synthesis of 1-aminoethenol, the higher-energy tautomer of acetamide, that has not been spectroscopically identified to date. The title compound was prepared by flash vacuum pyrolysis of malonamic acid and was characterized employing Received 4th September 2020 matrix isolation infrared as well as ultraviolet/visible spectroscopy. Coupled-cluster computations at the Accepted 19th October 2020 AE-CCSD(T)/cc-pVTZ level of theory support the spectroscopic assignments. Upon photolysis at l > DOI: 10.1039/d0sc04906a 270 nm, the enol rearranges to acetamide as well as ketene and ammonia. As the latter two are even rsc.li/chemical-science higher in energy, they constitute viable starting materials for formation of the title compound. Creative Commons Attribution 3.0 Unported Licence. Introduction molecules24 have been identied in the interstellar medium (ISM) through their rotational and vibrational transitions and Complex molecular structures typically form from thermody- there is evidence for the existence of larger structures that await 15,25 namically very stable and thus unreactive small molecules that their identication. The interstellar presence of (pre)biotic oen have to be transformed rst into more reactive isomers molecules suggests that these may have extraterrestrial origin 26 before they can react. -

Synthesis of Acetonitrile by Dehydration of Acetamide
Indian Journal of Chemical Technology Yol. 5, November 1998, pp. 405-406 NOTE. Synthesis of acetonitrile by dehydration circulating ice cold water. The analysis of products of acetamide on an active ZnO catalyst: A was carried out on a Chemito 8610 Gas comparison with zeolite catalysts. chromatograph. Acetonitrile was analysed on SE ,30 column using FID detector whereas Sajo P Naik & Julio B Femandes· decomposition products from propan-2-01 were Department of Chemistry, Goa University, Talegao analysed on a CarboWax column using TCD Plateau, Goa, 403206, India detector. TPD of the catalyst was carried out using Received 25 November 1997; accepted 2 July 1998 NH3 as a probe molecule. Before carrying out adsorption of ammonia, the catalysts were Catalytic activities of three different zinc oxides is activated at 393 K in a. stream of oxygen gas. evaluated in dehydration of acetamide reaction at 723 K. Dried ammonia gas was then passed over the It is seen that an oxide of zinc, ZnO(u) prepared by catalyst for 30 min at a flow rate of 5 mUmin. decomposition of a mixture of zinc nitrate, urea and The amount of ammonia desorped at various oxalic acid method showed much higher activity than temperatures gave measure of catalyst acidity other zinc oxides. Also the activity of ZnO(u) was comparable with the reported activities of zeolite which was expressed in mmollg of catalyst. catalysts. The activity of the catalysts could be correlated with their acidity. Results and Discussion Activity data' for zinc oxides and some zeolite catalysts in dehydration reaction of acetamide are presented in Table 1, where as activity of zinc Organic nitriles are synthesised from a number of oxides4 in decomposition of propan-2-01 is reagentsl• The synthesis of nitriles from esters and presented in Table 2. -

Practical Preparatory Problem Icho 2018
19th – 29th July 2018 Bratislava, SLOVAKIA Prague, CZECH REPUBLIC www.50icho.eu PREPARATORY PROBLEMS: PRACTICAL 50th IChO 2018 International Chemistry Olympiad SLOVAKIA & CZECH REPUBLIC BACK TO WHERE IT ALL BEGAN UPDATED 1st JUNE 2018 INTERNATIONAL CHEMISTRY OLYMPIAD / SLOVAKIA & CZECH REPUBLIC, 2018 Table of Contents Contributing Authors ................................................................................................................... 2 Fields of Advanced Difficulty ...................................................................................................... 2 Safety ......................................................................................................................................... 3 Problem P1. Determination of a metallic ore composition ........................................................... 4 Problem P2. Determination of a carbonate rock composition ..................................................... 8 Problem P3. Determination and identification of organic acids ................................................. 11 Problem P4. A chemical oscillator and its activation energies................................................... 15 Problem P5. Kinetics of a chemical wave front propagation ..................................................... 19 Problem P6. Separation of acidic, basic and neutral organic compounds ................................. 22 Problem P7. Meerwein–Ponndorf–Verley reduction ................................................................. 26 Problem P8. Transformation -

The Process of Acetonitrile Synthesis Over Γ-Al2o3 Promoted By
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Electronic archive of Tomsk Polytechnic University Available online at www.sciencedirect.com ScienceDirect Procedia Chemistry 10 ( 2014 ) 108 – 113 XV International Scientific Conference “Chemistry and Chemical Engineering in XXI century” dedicated to Professor L.P. Kulyov The Process of Acetonitrile Synthesis over Ȗ-Al2O3 Promoted by Phosphoric Acid Catalysts ∗ Sergey I. Galanova , Olga I. Sidorovaa, Mikhail A. Gavrilenkob aTomsk State University, 36, Lenin Ave., 634050, Tomsk, Russia bTomsk Polytechnic University, 30, Lenin Ave., 634050, Tomsk, Russia Abstract The influence of principal parameters (reaction temperature, ratio of acetic acid and ammonia, composition of reactionary mixture and promotion of catalysts) on the selectivity and yield of the desired product was studied in the reaction of catalytic acetonitrile synthesis by ammonolysis of acetic acid. The processing of Ȗ-Al2O3 by phosphoric acid increases amount of the centers, on which carries out reaction of acetamide dehydration. The kinetic model of a limiting stage of reaction – the acetamide dehydration to acetonitrile was suggested. In the process of ammonolysis of acetic acid it was demonstrated that the use of catalysts promoted by phosphoric acid and ratio NH3:CH3COOH=(3-4):1 at temperatures of a reactor 360-390 °ɋ leads to the increase of acetonitrile productivity to 0.7-0.8 g/cm3·h and allows to minimize formation of by-products. ©© 20142014 Published The Authors. by Elsevier Published B.V. byThis Elsevier is an open B.V. access article under the CC BY-NC-ND license (Peer-reviewhttp://creativecommons.org/licenses/by-nc-nd/3.0/ under responsibility of Tomsk Polytechnic). -

United States Patent (19) (11 Patent Number: 4,918,222 Lin Et Al
United States Patent (19) (11 Patent Number: 4,918,222 Lin et al. 45 Date of Patent: Apr. 17, 1990 (54) PROCESS FOR SYNTHESIS OF (56) References Cited N-ACETYLGLYCINE U.S. PATENT DOCUMENTS (75) Inventors: Jiang-Jen Lin, Round Rock; John F. 3,213,155 10/1965 Schriesheim et al. .............. 562/544 Knifton; Ernest L. Yeakey, both of . 3,766,266 10/1973 Wakamatsu et al. ............... 562/518 Austin, all of Tex. FOREIGN PATENT DOCUMENTS 73 Assignee: Texaco Inc., White Plains, N.Y. 1326014 8/1973 United Kingdom ................ 562/519 Primary Examiner-Vivian Garner 21 Appl. No.: 83,397 Attorney, Agent, or Firm-Jack H. Park; Kenneth R. 22) Filed: Aug. 10, 1987 Priem; Cynthia L. Kendrick (57) ABSTRACT Related U.S. Application Data An N-acetylglycine is manufactured by reacting para (63) Continuation-in-part of Ser. No. 635,077, Jul. 27, 1984, formaldehyde with an acetamide and carbon monoxide abandoned. in the present of a cobalt-containing catalyst promoted by a sulfoxide or dinitride compounds. The presence of (51) Int. Cl. .................. C07C102/00; C07C 103/50; sulfoxide or dinitrile ligands are essential for the high CO7C 103/48 yield synthesis of N-acetylglycine and good cobalt re 52) U.S. C. .................................... 562/518; 564/132; covery. 564/159 (58) Field of Search ................................ 562/518, 575 1 Claim, No Drawings 4,918,222 1. 2 N-acetylglycine was not encountered by other amidoa PROCESS FOR SYNTHESIS OF cid analogs. N-ACETYLGLYCNE N-acetylglycine (the smallest molecule in the amido acid family) has a melting point of 207 C. -

INTERMEDIATE CHEMICALS for DYES APPENDIX Harmonized Tariff Schedule of the United States (2009) Annotated for Statistical Reporting Purposes
Harmonized Tariff Schedule of the United States (2009) Annotated for Statistical Reporting Purposes INTERMEDIATE CHEMICALS FOR DYES APPENDIX Harmonized Tariff Schedule of the United States (2009) Annotated for Statistical Reporting Purposes INTERMEDIATE CHEMICALS FOR DYES APPENDIX 2 This appendix enumerates those intermediate chemicals for dyes which are eligible for duty-free treatment under the provisions of general note 14 of the tariff schedule. Product CAS Number Acetaldehyde, (1,3-dihydro-1,3,3-trimethyl-2H-indol-2-ylidene)- ............................................... 84-83-3 Acetamide, N-(3-amino-4-methoxyphenyl)- ............................................................. 6375-47-9 Acetamide, N-(3-aminophenyl)- ....................................................................... 102-28-3 Acetamide, N-(4-aminophenyl)- ....................................................................... 122-80-5 Acetamide, N-(3-aminophenyl)-, monohydrochloride ...................................................... 621-35-2 Acetamide, N-(4-aminophenyl)-N-methyl- ............................................................... 119-63-1 Acetamide, N-(2,5-dimethoxy phenyl)- ................................................................ 3467-59-2 Acetamide, N-(7-hydroxy-1-naphthalenyl- .............................................................. 6470-18-4 Acetamide, N-(2-methoxy-5-methyl phenyl)- ............................................................ 6962-44-3 Acetamide, N-(2-Methoxy Phenyl)- .................................................................... -
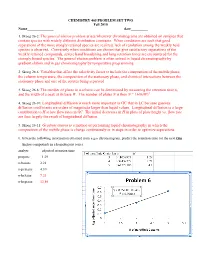
Problem Set Two F'09-Solution
CHEMISTRY 465 PROBLEM SET TWO Fall 2010 Name date 1. Skoog 26-2: The general elution problem arises whenever chromatograms are obtained on samples that contain species with widely different distribution constants. When conditions are such that good separations of the more stongly retained species are realized, lack of resolution among the weakly held species is observed. Conversely when conditions are chosen that give satisfactory separations of the weakly retained compounds, severe band broadening and long retention times are encountered for the strongly bound species. The general elution problem is often solved in liquid chromatography by gradient elution and in gas chromatography by temperature programming 2. Skoog 26-6: Variables that affect the selectivity factor α include the composition of the mobile phase, the column temperature, the composition of the stationary phase, and chemical interactions between the stationary phase and one of the solutes being separated 3. Skoog 26-8: The number of plates in a column can be determined by measuring the retention time tR 2 and the width of a peak at its base W. The number of plates N is then N = 16(tR/W) 4. Skoog 26-10: Longitudinal diffusion is much more important in GC that in LC because gaseous diffusion coefficients are orders of magnitude larger than liquid values. Longitudinal diffusion is a large contribution to H at low flow rates in GC. The initial decreases in H in plots of plate height vs. flow rate are thus largely the result of longitudinal diffusion 5. Skoog 26-11: Gradient elution is a method of performing liquid chromatography in which the composition of the mobile phase is change continuously or in steps in order to optimize separations. -

Chemical Reactions in the Nitrogen-Acetone Ice Induced by Cosmic Ray
Chemical reactions in the nitrogen-acetone ice induced by cosmic ray analogs: relevance for the Solar System A. L. F. de Barros1,2, D. P. P. Andrade3, E. F. da Silveira4, K. F. Alcantara4, 5 5 P. Boduch , H. Rothard 1 Departamento de F´ısica, Centro Federal de Educa¸c˜ao Tecnolo´gica Celso Suckow da Fonseca, Av. Maracana˜ 229, 20271-110 Rio de Janeiro, RJ, Brazil 2 NASA Ames Research Center, Mail Stop 245-6, Moffett Field, CA 94035-1000, USA 3 Universidade Federal do Rio de Janeiro, Observatorio do Valon go, Ladeira Pedro Antonio, 43, Rio de Janeiro, Brazil 4 Departamento de F´ısica, Pontif´ıcia Universidade Cat´olica do Rio de Janeiro, Rua Marquˆes de S˜ao Vicente 225, 22451-900, Rio de Janeiro, RJ, Brazil 5 Centre de Recherche sur les Ions, les Mat´eriaux et la Photonique Normandie Univ, ENSICAEN, UNICAEN, CEA, CNRS, CIMAP, 14000 Caen, France ABSTRACT The radiolysis of a 10:1 nitrogen:acetone mixture, condensed at 11 K, by 40 MeV 58Ni11+ ions is studied. These results are representative of studies concerning solar system objects exposed to cosmic rays. In the Kuiper Belt, region of Trans-Neptunian Objects (TNOs), acetone, N2 and other small molecules were detected and may be present on icy surfaces. Bombardment by cosmic rays triggers chemical reactions leading to synthesis of larger molecules. In this work, destruction cross sections of acetone and nitrogen molecules in solid phase are determined from a sequence of infrared spectra obtained at increasing ion beam fluence. The results are analyzed and compared with those of previous experiments performed with pure acetone.