Chapter 1: Introduction
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Reigniting Pharmaceutical Innovation Through Holistic Drug Targeting
Drug Discovery Reigniting pharmaceutical innovation through holistic drug targeting Modern drug discovery approaches take too long, are too expensive, have too many clinical failures and uncertain outcomes. There are many reasons for this unsustainable business model, but primarily, the approaches are not comprehensively holistic. Secondly, none of the pharmaceutical companies openly share the reasons for the failure of their clinical candidates in real time to effectively navigate the ‘industry’ from committing the same mistakes. It is time for the pharmaceutical industry to embrace, metaphorically speaking, a community-driven ‘Wikipedia’ or ‘Waze’-type shared-knowledge, openly- accessible innovation model to harvest data and create a crowd-sourced path towards a safer and faster road to the discovery and development of life-saving medicines. This may be a bitter pill for Pharma to swallow, but one that ought to be given serious consideration. The time is now for a paradigm shift towards multi-target-network polypharmacology drugs exalting symphonic or concert performance with occasional soloists to reignite pharmaceutical innovation. rom the turn of the 20th century, pharma- and ultra-high throughput workflows enabled By Dr Anuradha Roy, cognosy and ethnopharmacology combined screening of millions of compounds to identify hit Professor Bhushan F with anecdotal clinical evidence accumulat- can didates for lead development. Despite billions Patwardhan and ed over centuries of hands-on knowledge from pri- of dollars spent on R&D, only a fraction of the Dr Rathnam mordial disease management practices, albeit with molecules identified from the screening operations Chaguturu uncertain outcomes, formed the basis for the devel- find their way into clinical trials. -

Development of a FLIPR Assay for the Simultaneous Identification of Mrgd
Hindawi Publishing Corporation Journal of Biomedicine and Biotechnology Volume 2010, Article ID 326020, 8 pages doi:10.1155/2010/326020 Research Article Development of a FLIPR Assay for the Simultaneous Identification of MrgD Agonists and Antagonists from a Single Screen Seena K. Ajit,1, 2 Mark H. Pausch,2 Jeffrey D. Kennedy,2 and Edward J. Kaftan2 1 Department of Pharmacology and Physiology, Drexel University College of Medicine, 245 North 15th Street, MS number 488, Philadelphia, PA 19102, USA 2 Neuroscience Discovery, Pfizer Global Research and Development, CN 8000, Princeton, NJ 08543, USA Correspondence should be addressed to Seena K. Ajit, [email protected] Received 1 April 2010; Revised 8 July 2010; Accepted 6 August 2010 Academic Editor: John V. Moran Copyright © 2010 Seena K. Ajit et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. MrgD, a member of the Mas-related gene family, is expressed exclusively in small diameter IB4+ neurons in the dorsal root ganglion. This unique expression pattern, the presence of a single copy of MrgD in rodents and humans, and the identification of a putative ligand, beta-alanine, make it an experimentally attractive therapeutic target for pain with limited likelihood of side effects. We have devised a high throughput calcium mobilization assay that enables identification of both agonists and antagonists from a single screen for MrgD. Screening of the Library of Pharmacologically Active Compounds (LOPAC) validated this assay approach, and we identified both agonists and antagonists active at micromolar concentrations in MrgD expressing but not in parental CHO-DUKX cell line. -

Innovative Approaches in Drug Discovery
See discussions, stats, and author profiles for this publication at: https://www.researchgate.net/publication/313600306 Reverse pharmacology and system approach for drug discovery and development Article · January 2008 CITATIONS READS 9 428 4 authors: Bhushan K Patwardhan Ashok D B Vaidya Savitribai Phule Pune University Saurashtra University 229 PUBLICATIONS 5,729 CITATIONS 237 PUBLICATIONS 2,356 CITATIONS SEE PROFILE SEE PROFILE Mukund S Chorghade Swati P Joshi THINQ Pharma and Empiriko CSIR - National Chemical Laboratory, Pune 117 PUBLICATIONS 1,428 CITATIONS 59 PUBLICATIONS 541 CITATIONS SEE PROFILE SEE PROFILE Some of the authors of this publication are also working on these related projects: graduate studies View project Standardization of an Ayurveda-inspired antidiabetic and experimental studie with state-of--the artinvitro and in vivo models, View project All content following this page was uploaded by Mukund S Chorghade on 24 May 2017. The user has requested enhancement of the downloaded file. Chapter 4 Reverse Pharmacology Ashwinikumar A. Raut1, Mukund S. Chorghade2 and Ashok D.B. Vaidya1 1Kasturba Health Society-Medical Research Centre, Mumbai, Maharashtra, India, 2THINQ, Boston, MA, United States INTRODUCTION AND BACKGROUND I never found it [drug discovery] easy. People say I was lucky twice but I resent that. We stuck with [cimetidine] for 4 years with no progress until we eventually succeeded. It was not luck, it was bloody hard work. — Sir James Black, Nobel Laureate (Jack, 2009). Introduction The aforementioned quote from Sir James Black, the discoverer of β-adrenergic and H2- blockers, expresses the exasperation so often felt by many scientists who have dedicated their lives to new drug discoveries. -

Application of Multi-Target Phytotherapeutic Concept in Malaria
Tarkang et al. Biomarker Research (2016) 4:25 https://doi.org/10.1186/s40364-016-0077-0 REVIEW Open Access Application of multi-target phytotherapeutic concept in malaria drug discovery: a systems biology approach in biomarker identification Protus Arrey Tarkang1,2*, Regina Appiah-Opong2, Michael F. Ofori3, Lawrence S. Ayong4 and Alexander K. Nyarko2,5 Abstract There is an urgent need for new anti-malaria drugs with broad therapeutic potential and novel mode of action, for effective treatment and to overcome emerging drug resistance. Plant-derived anti-malarials remain a significant source of bioactive molecules in this regard. The multicomponent formulation forms the basis of phytotherapy. Mechanistic reasons for the poly- pharmacological effects of plants constitute increased bioavailability, interference with cellular transport processes, activation of pro-drugs/deactivation of active compounds to inactive metabolites and action of synergistic partners at different points of the same signaling cascade. These effects are known as the multi-target concept. However, due to the intrinsic complexity of natural products-based drug discovery, there is need to rethink the approaches toward understanding their therapeutic effect. This review discusses the multi-target phytotherapeutic concept and its application in biomarker identification using the modified reverse pharmacology - systems biology approach. Considerations include the generation of a product library, high throughput screening (HTS) techniques for efficacy and interaction assessment, High Performance Liquid Chromatography (HPLC)-based anti-malarial profiling and animal pharmacology. This approach is an integrated interdisciplinary implementation of tailored technology platforms coupled to miniaturized biological assays, to track and characterize the multi-target bioactive components of botanicals as well as identify potential biomarkers. -

Current Screening Methodologies in Drug Discovery for Selected Human Diseases
marine drugs Review Current Screening Methodologies in Drug Discovery for Selected Human Diseases Olga Maria Lage 1,2,*, María C. Ramos 3, Rita Calisto 1,2, Eduarda Almeida 1,2, Vitor Vasconcelos 1,2 ID and Francisca Vicente 3 1 Departamento de Biologia, Faculdade de Ciências, Universidade do Porto, Rua do Campo Alegre s/nº 4169-007 Porto, Portugal; [email protected] (R.C.); [email protected] (E.A.); [email protected] (V.V.) 2 CIIMAR/CIMAR–Centro Interdisciplinar de Investigação Marinha e Ambiental–Universidade do Porto, Terminal de Cruzeiros do Porto de Leixões, Avenida General Norton de Matos, S/N, 4450-208 Matosinhos, Portugal 3 Fundación MEDINA, Centro de Excelencia en Investigación de Medicamentos Innovadores en Andalucía, Parque Tecnológico de Ciencias de la Salud, 18016 Granada, Spain; [email protected] (M.C.R.); [email protected] (F.V.) * Correspondence: [email protected]; Tel.: +351-22-0402724; Fax.: +351-22-0402799 Received: 7 August 2018; Accepted: 11 August 2018; Published: 14 August 2018 Abstract: The increase of many deadly diseases like infections by multidrug-resistant bacteria implies re-inventing the wheel on drug discovery. A better comprehension of the metabolisms and regulation of diseases, the increase in knowledge based on the study of disease-born microorganisms’ genomes, the development of more representative disease models and improvement of techniques, technologies, and computation applied to biology are advances that will foster drug discovery in upcoming years. In this paper, several aspects of current methodologies for drug discovery of antibacterial and antifungals, anti-tropical diseases, antibiofilm and antiquorum sensing, anticancer and neuroprotectors are considered. -

Anew Drug Design Strategy in the Liht of Molecular Hybridization Concept
www.ijcrt.org © 2020 IJCRT | Volume 8, Issue 12 December 2020 | ISSN: 2320-2882 “Drug Design strategy and chemical process maximization in the light of Molecular Hybridization Concept.” Subhasis Basu, Ph D Registration No: VB 1198 of 2018-2019. Department Of Chemistry, Visva-Bharati University A Draft Thesis is submitted for the partial fulfilment of PhD in Chemistry Thesis/Degree proceeding. DECLARATION I Certify that a. The Work contained in this thesis is original and has been done by me under the guidance of my supervisor. b. The work has not been submitted to any other Institute for any degree or diploma. c. I have followed the guidelines provided by the Institute in preparing the thesis. d. I have conformed to the norms and guidelines given in the Ethical Code of Conduct of the Institute. e. Whenever I have used materials (data, theoretical analysis, figures and text) from other sources, I have given due credit to them by citing them in the text of the thesis and giving their details in the references. Further, I have taken permission from the copyright owners of the sources, whenever necessary. IJCRT2012039 International Journal of Creative Research Thoughts (IJCRT) www.ijcrt.org 284 www.ijcrt.org © 2020 IJCRT | Volume 8, Issue 12 December 2020 | ISSN: 2320-2882 f. Whenever I have quoted written materials from other sources I have put them under quotation marks and given due credit to the sources by citing them and giving required details in the references. (Subhasis Basu) ACKNOWLEDGEMENT This preface is to extend an appreciation to all those individuals who with their generous co- operation guided us in every aspect to make this design and drawing successful. -
Course Syllabus - TRBIO 450
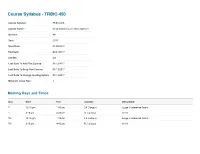
Course Syllabus - TRBIO 450 Course Number: TRBIO 450 Course Name: Drug Discovery and Development Quarter: WI Year: 2017 Start Date: 01/05/2017 End Date: 03/24/2017 Credits: 3.0 Last Date To Add This Course: 01/13/2017 Last Date To Drop This Course: 01/13/2017 Last Date To Change Grading Option: 01/13/2017 Minimum Class Size: 1 Meeting Days and Times Day Start End Location Description T 12:15 pm 1:45 pm CA Campus Large Conference Room T 3:15 pm 4:45 pm FL Campus A116 TH 12:15 pm 1:45 pm CA Campus Large Conference Room TH 3:15 pm 4:45 pm FL Campus A116 Course Managers First Mail Organization Name (non-TSRI Role Last Name Department Phone Email Name Code personnel) Course Department of Molecular (561) 228- McDonald Patricia 2A2 [email protected] Director Therapeutics 2222 Course Department of Molecular (561) 228- Duckett Derek 2A2 [email protected] Director Therapeutics 2224 Department of Molecular (561) 228- Admin Clark Pamela 2A2 [email protected] Therapeutics 2029 Nieto Department of Molecular (561) 228- TA Ainhoa 2A1 [email protected] Gutierrez Therapeutics 2856 (858) 784- TA Vaughan Megan TSRI Graduate Program TPC-19 [email protected] 8469 Course Description The course reviews the processes through which potential new therapeutics are identified. This includes the description of the roles played by a wide range of scientific disciplines, including biology, chemistry, in vitro/in vivo pharmacology, drug metabolism, pharmacokinetics, and ADME/Tox in the early drug discovery process. The complexity of ddiscovering drugs that may be a commercial success, or a public health success, involves a complex interaction between investors, industry, academia, patent laws, regulatory exclusivity, marketing and the need to balance secrecy with communication will be reviewed. -

Drug Development
DRUG DEVELOPMENT www.acuteleuk.org Drug Development Process Only about 2% of substances evaluated in early research make it to the market as new medicines Number of drugs in the R&D pipeline Number of drugs in the R&D pipeline worldwide 2019 vs. 2020, by development phase Source: https://www.statista.com/ A minimum of a 10-year plan It takes over 10 years and on average costs between €400 million and €1.5 billion before a new medicine can be made available to patients and reimbursement starts Drug development means … What is the rationale to initiate drug development ? • Scientific and Intellectual Property – How does the drug work? • Molecule structure and properties • Mechanism of action • Biological activity (bioassays, drug/target interaction, affinity, specificity,…) – What data support the approach? • Literature validation • In-house preclinical/clinical data – Who owns the intellectual property? What is the rationale to initiate drug development ? • Disease overview and unmet medical need – What is known about disease pathogenesis (translational medicine)? – Where in the disease process should drug enter? Greatest unmet need? – Is this disease part of a disease family? If so which fits best with drug and regulatory history? Is there an orphan approach? – Is this disease field crowded? – Has the indication been clinically studied before (in drug studies)? – What is the event rate for target medical problems? (Make the statisticians happy) – Are there “approvable” endpoints for the unmet needs? – If not, what endpoints seem reasonable: -

Reverse Pharmacology-A Paradigm Shift for Drug Discovery and Development
Current Research in Drug Discovery Original Research Paper Reverse Pharmacology-A Paradigm Shift for Drug Discovery and Development Ashok D.B. Vaidya Kasturba Health Society, Advanced Centre of Reverse Pharmacology (ICMR) 17, Khandubhai Desai Road, Vile Parle (West), Mumbai, 400 056, India Department of Pharmaceutical Sciences, Saurashtra University, India Article history Abstract: The current scenario of drug discovery and development is Received: 06-11-2014 facing a model perturbation, despite significant advances in genomics, Revised: 28-11-2014 proteomics, metabolomics, combinatorial chemistry and high through put Accepted: 05-12-2014 assays. There is an urgent need of a paradigm shift in approaches to drug discovery. Natural products of Ayurveda offer a vast potential for novel Corresponding Author: phytomolecules with clinical activity. In India, Ayurveda is availed of by Ashok D.B. Vaidya Kasturba Health Society, more than 70% of the population. There has been a renaissance in scientific Advanced Centre of Reverse exploration of medicinal plants with therapeutic activity. New methods have Pharmacology (ICMR) 17, been proposed and developed for such exploration. Ayurvedic Khandubhai Desai Road, Vile Pharmacoepidemiology, Observational therapeutics and Reverse Parle (West), Pharmacology paths have led to significant hits, leads and drug candidates for Mumbai, 400 056, India several diseases. The active phyto-molecules will also provide novel Email: [email protected] scaffolds for medicinal chemists to enhance efficacy and reduce toxicity. Several plants have shown promise Viz. Mucuna pruriens for Parkinson’s disease, Nycthanthes arbor-tritis for malaria and Phyllanthus amarus for hepatitis. This approach of Reverse Pharmacology has also been adopted globally by several groups. A plea is made to look at the therapeutic activity of natural products for a revolution in drug discovery and development. -

Chapter 11. the Drug Discovery Process
Chapter 11 The Drug Discovery Process Success is the ability to go from failure to failure with no lack of enthusiasm... — Winston Churchill (1874À1965) I am interested in physical medicine because my father was. I am interested in medical research because I believe in it. I am interested in arthritis because I have it... — Bernard Baruch, 1959 ...new techniques may be generating bigger haystacks as opposed to more needles ... — D.F. Horrobin, 2000 11.1 Some Challenges for Modern Drug 11.5 Types of Therapeutically Active 11.8 Pharmacodynamics and High- Discovery Ligands: Polypharmacology Throughput Screening 11.2 Target-Based Drug Discovery 11.6 Pharmacology in Drug Discovery 11.9 Drug Development 11.3 Systems-Based Drug Discovery 11.7 Chemical Sources for Potential 11.10 Clinical Testing 11.4 In vivo Systems, Biomarkers, and Drugs 11.11 Summary and Conclusions Clinical Feedback References 11.1 SOME CHALLENGES FOR MODERN robotic screening using simplistic single gene target DRUG DISCOVERY approaches (inappropriate reliance on the genome as an instruction booklet for new drugs) coupled with a de- The identification of primary biological activity at the tar- emphasis of pharmacological training may have combined get of interest is just one of a series of requirements for a to cause the current deficit in new drugs [2]. The lack of drug. The capability to screen massive numbers of com- success in drug discovery is reflected in the number of ELSEVIERÀ drugs that have failed in the transition from Phase II clini- pounds has increased dramatically over the past 10 15 years, yet no corresponding increase in successfully cal trials (trial in a small number of patients designed to launched drugs has ensued. -

IJP October 06.Pmd
Special Article Reverse pharmacological correlates of ayurvedic drug actions*actions* A.D.B. Vaidya Bhavan’s Swami Prakashanand Ayurvedic Research Center (SPARC), Mumbai – 400 049. India Received: 28.7.2005 Revised: 22.5.2006 Accepted: 30.5.2006 Correspondence to: Ashok DB Vaidya Sir Ram Nath Chopra E-mail: [email protected] The human condition has been enhanced considerably by While Sir Ram Nath Chopra and other West-trained those biochemists and pharmacologists who have applied the scientists were evaluating the effects of ayurvedic drugs and scientific method to prove ancient myths and legends. For plant extracts on tissues and animals, eminent vaidyas such example, the mystical powers of the snakeroot plant of India, as Mahamahopadhyaya Gananath Sen were laying the either ignored or ridiculed by scientists for decades, were foundation of ‘Reverse Pharmacology’. The science of dispelled with the isolation and identification of its active integrating documented clinical/experiential hits into leads by transdisciplinary exploratory studies, reverse alkaloid. pharmacology further developed these leads into drug candidates by experimental and clinical research. The – Richard E Davis proposed approach is the new paradigm or a rediscovered paradigm. “Rediscovered” because, even in modern Sir Ram Nath Chopra (1882-1973) was a pioneer in the pharmacology, some of the major fundamental discoveries field of experimental pharmacology of indigenous drugs of evolved from plant effects in humans. Table 1 lists some India. Most of the medicinal plants he studied were in use, in such historical examples of reverse pharmacology. The animal ayurveda, for thousands of years. At the Calcutta School of models and/or mechanisms of such human effects often Tropical Medicine, he pioneered a paradigm, a pace and a provided the foundation of pharmacology. -

Reverse Pharmacology and Drug Discovery: Artemisia Annua and Its Anti-HIV Activity
Chapter 14 Reverse Pharmacology and Drug Discovery: Artemisia annua and Its Anti-HIV Activity Frank van der Kooy 14.1 Introduction 14.1.1 Drug Discovery and Reverse Pharmacology There are various ways in which new drugs can be developed. One approach is in silico drug design based on our existing knowledge of the biology of a specific disease and the specific target site binding chemistry. Based on this knowledge, a range of molecules will be designed and synthesised after which they will be tested in in vitro bioassays for activity and toxicity. The best candidates, called lead compounds, will then be ‘‘fine-tuned’’by chemical derivatisation in order to improve their activity and/or to reduce their toxicity. Lead compounds are then tested in various animal models before entering clinical trials in people. Another approach is to screen a large number of biological samples (plants, bacteria and fungi) for activity against a specific disease. Any active extract, consisting of many compounds, will be fractionated by chromatographic techniques, and each fraction will be tested for in vitro activity. Active fractions will again be fractionated until the active compound is identified. This process, also called bioguided fractionation, can go through a number of fractionation cycles before the active compound is identified. The active compound will be chemically derivatised in order to improve its properties before in vivo animal studies will be conducted. Based on these test results, the most promising lead compounds will then be tested in clinical trials in people. There are however a number of shortcomings with both approaches.