Summary Review
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Picrotoxin-Like Channel Blockers of GABAA Receptors
COMMENTARY Picrotoxin-like channel blockers of GABAA receptors Richard W. Olsen* Department of Molecular and Medical Pharmacology, Geffen School of Medicine, University of California, Los Angeles, CA 90095-1735 icrotoxin (PTX) is the prototypic vous system. Instead of an acetylcholine antagonist of GABAA receptors (ACh) target, the cage convulsants are (GABARs), the primary media- noncompetitive GABAR antagonists act- tors of inhibitory neurotransmis- ing at the PTX site: they inhibit GABAR Psion (rapid and tonic) in the nervous currents and synapses in mammalian neu- system. Picrotoxinin (Fig. 1A), the active rons and inhibit [3H]dihydropicrotoxinin ingredient in this plant convulsant, struc- binding to GABAR sites in brain mem- turally does not resemble GABA, a sim- branes (7, 9). A potent example, t-butyl ple, small amino acid, but it is a polycylic bicyclophosphorothionate, is a major re- compound with no nitrogen atom. The search tool used to assay GABARs by compound somehow prevents ion flow radio-ligand binding (10). through the chloride channel activated by This drug target appears to be the site GABA in the GABAR, a member of the of action of the experimental convulsant cys-loop, ligand-gated ion channel super- pentylenetetrazol (1, 4) and numerous family. Unlike the competitive GABAR polychlorinated hydrocarbon insecticides, antagonist bicuculline, PTX is clearly a including dieldrin, lindane, and fipronil, noncompetitive antagonist (NCA), acting compounds that have been applied in not at the GABA recognition site but per- huge amounts to the environment with haps within the ion channel. Thus PTX major agricultural economic impact (2). ͞ appears to be an excellent example of al- Some of the other potent toxicants insec- losteric modulation, which is extremely ticides were also radiolabeled and used to important in protein function in general characterize receptor action, allowing and especially for GABAR (1). -

The Anxiomimetic Properties of Pentylenetetrazol in the Rat
University of Rhode Island DigitalCommons@URI Open Access Dissertations 1980 THE ANXIOMIMETIC PROPERTIES OF PENTYLENETETRAZOL IN THE RAT Gary Terence Shearman University of Rhode Island Follow this and additional works at: https://digitalcommons.uri.edu/oa_diss Recommended Citation Shearman, Gary Terence, "THE ANXIOMIMETIC PROPERTIES OF PENTYLENETETRAZOL IN THE RAT" (1980). Open Access Dissertations. Paper 165. https://digitalcommons.uri.edu/oa_diss/165 This Dissertation is brought to you for free and open access by DigitalCommons@URI. It has been accepted for inclusion in Open Access Dissertations by an authorized administrator of DigitalCommons@URI. For more information, please contact [email protected]. THE ANXIOMIMETIC PROPERTIES OF PENTYLENETETRAZOL IN THE RAT BY GARY TERENCE SHEARMAN A DISSERTATION SUBMITTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY IN PHARMACEUTICAL SCIENCES (PHARMACOLOGY AND TOXICOLOGY) UNIVERSITY OF RHODE ISLAND 19 80 DOCTOR OF PHILOSOPHY DISSERT.A.TION OF GARY TERENCE SHEAffiil.AN Approved: Dissertation Cormnittee \\ Major Professor ~~-L_-_._dd__· _... _______ _ -~ar- Dean of the Graduate School UNIVERSITY OF RHODE ISLAND 1980 ABSTRACT Investigation of the biological basis of anxiety is ham pered by the lack of an appropriate animal model for research purposes. There are no known drugs that cause anxiety in laboratory animals. Pentylenetetrazol (PTZ) produces intense anxiety in human volunteers (Rodin, 1958; Rodin and Calhoun, 1970). Therefore, it was the major objective of this disser- tation to test the hypothesis that the discriminative stimu lus produced by PTZ in the rat was related to its anxiogenic action in man. It was also an objective to suggest the neuro- chemical basis for the discriminative stimulus property of PTZ through appropriate drug interactions. -

TRIDIONE® (Trimethadione) Tablets
TRIDIONE® (trimethadione) Tablets BECAUSE OF ITS POTENTIAL TO PRODUCE FETAL MALFORMATIONS AND SERIOUS SIDE EFFECTS, TRIDIONE (trimethadione) SHOULD ONLY BE UTILIZED WHEN OTHER LESS TOXIC DRUGS HAVE BEEN FOUND INEFFECTIVE IN CONTROLLING PETIT MAL SEIZURES. DESCRIPTION TRIDIONE (trimethadione) is an antiepileptic agent. An oxazolidinedione compound, it is chemically identified as 3,5,5-trimethyloxozolidine-2,4-dione, and has the following structural formula: TRIDIONE is a synthetic, water-soluble, white, crystalline powder. It is supplied in tablets for oral use only. Inactive Ingredients 150 mg Dulcet Tablet: Corn starch, lactose, magnesium stearate, magnesium trisilicate, sucrose and natural/synthetic flavor. CLINICAL PHARMACOLOGY TRIDIONE has been shown to prevent pentylenetetrazol-induced and thujone-induced seizures in experimental animals; the drug has a less marked effect on seizures induced by picrotoxin, procaine, cocaine, or strychnine. Unlike the hydantoins and antiepileptic barbiturates, TRIDIONE does not modify the maximal seizure pattern in patients undergoing electroconvulsive therapy. TRIDIONE has a sedative effect that may increase to the point of ataxia when excessive doses are used. A toxic dose of the drug in animals (approximately 2 g/kg) produced sleep, unconsciousness, and respiratory depression. Trimethadione is rapidly absorbed from the gastrointestinal tract. It is demethylated by liver microsomes to the active metabolite, dimethadione. Approximately 3% of a daily dose of TRIDIONE is recovered in the urine as unchanged drug. The majority of trimethadione is excreted slowly by the kidney in the form of dimethadione. INDICATIONS TRIDIONE (trimethadione) is indicated for the control of petit mal seizures that are refractory to treatment with other drugs. CONTRAINDICATIONS TRIDIONE is contraindicated in patients with a known hypersensitivity to the drug. -

Pentetrazol for Idiopathic Hypersomnia and Narcolepsy Type 2 NIHRIO (HSRIC) ID: 11738 NICE ID: 9624
NIHR Innovation Observatory Evidence Briefing: February 2018 Pentetrazol for idiopathic hypersomnia and narcolepsy type 2 NIHRIO (HSRIC) ID: 11738 NICE ID: 9624 LAY SUMMARY Hypersomnia means excessive sleepiness and sleeping, when the person struggles to stay awake during the day and has great difficulty being awakened from sleep. When there is no clear cause, this condition is called idiopathic hypersomnia (IH). Narcolepsy is a rare long-term brain disorder that causes a person to suddenly fall asleep at inappropriate times. Sometimes narcolepsy is associated with temporary loss of muscle control that causes weakness and possible collapse (cataplexy). This type of narcolepsy is called Type 1 narcolepsy. When narcolepsy is not associated with cataplexy is called type 2 narcolepsy. Narcolepsy and IH are very similar conditions. Pentetrazol is a medicinal product that is being developed for the treatment of IH and narcolepsy type 2. Pentetrazol is administered orally and it acts by blocking the effect of a chemical substance called Gamma-Aminobutyric Acid (GABA). GABA is thought to play a role in promoting sleeping and is believed to be elevated in people with IH. If licensed, Pentetrazol will offer a new treatment option for patients with IH or narcolepsy type 2. This briefing reflects the evidence available at the time of writing. A version of the briefing was sent to the company for a factual accuracy check. The company was unavailable to provide comment. It is not intended to be a definitive statement on the safety, efficacy or effectiveness of the health technology covered and should not be used for commercial purposes or commissioning without additional information. -

Chronic Intermittent Ethanol-Induced Switch of Ethanol Actions from Extrasynaptic to Synaptic Hippocampal
The Journal of Neuroscience, February 8, 2006 • 26(6):1749–1758 • 1749 Neurobiology of Disease Chronic Intermittent Ethanol-Induced Switch of Ethanol Actions from Extrasynaptic to Synaptic Hippocampal GABAA Receptors Jing Liang,1,2 Nianhui Zhang,3 Elisabetta Cagetti,2 Carolyn R. Houser,3 Richard W. Olsen,2 and Igor Spigelman1 1Division of Oral Biology and Medicine, School of Dentistry, University of California, Los Angeles, and Departments of 2Molecular and Medical Pharmacology and 3Neurobiology, David Geffen School of Medicine, University of California, Los Angeles, Los Angeles, California 90095 Alcohol withdrawal syndrome (AWS) symptoms include hyperexcitability, anxiety, and sleep disorders. Chronic intermittent ethanol (CIE) treatment of rats with subsequent withdrawal of ethanol (EtOH) reproduced AWS symptoms in behavioral assays, which included tolerance to the sleep-inducing effect of acute EtOH and its maintained anxiolytic effect. Electrophysiological assays demonstrated a CIE-induced long-term loss of extrasynaptic GABAA receptor (GABAAR) responsiveness and a gain of synaptic GABAAR responsiveness of CA1 pyramidal and dentate granule neurons to EtOH that we were able to relate to behavioral effects. After CIE treatment, the ␣4 3ϩ subunit-preferring GABAAR ligands 4,5,6,7 tetrahydroisoxazolo[5,4-c]pyridin-3-ol, La , and Ro15-4513 (ethyl-8-azido-5,6-dihydro-5- methyl-6-oxo-4H-imidazo[1,5␣][1,4]benzodiazepine-3-carboxylate) exerted decreased effects on extrasynaptic currents but had in- creased effects on synaptic currents. Electron microscopy revealed an increase in central synaptic localization of ␣4 but not ␦ subunits within GABAergic synapses on the dentate granule cells of CIE rats. Recordings in dentate granule cells from ␦ subunit-deficient mice revealed that this subunit is not required for synaptic GABAAR sensitivity to low [EtOH]. -

Cogentin (Benztropine Mesylate) Injection Label
COGENTIN® (benztropine mesylate injection) Rx Only DESCRIPTION Benztropine mesylate is a synthetic compound containing structural features found in atropine and diphenhydramine. It is designated chemically as 8-azabicyclo[3.2.1] octane, 3-(diphenylmethoxy)-,endo, methanesulfonate. Its empirical formula is C21H25NO•CH4O3S, and its structural formula is: Benztropine mesylate is a crystalline white powder, very soluble in water, and has a molecular weight of 403.54. COGENTIN (benztropine mesylate) is supplied as a sterile injection for intravenous and intramuscular use. Each milliliter of the injection contains: Benztropine mesylate…………………………………………………………………..1 mg Sodium chloride……………………………………………………….........................9 mg Water for injection q.s…………………………………………………………………..1 mL ACTIONS COGENTIN possesses both anticholinergic and antihistaminic effects, although only the former have been established as therapeutically significant in the management of parkinsonism. Page 1 of 7 Reference ID: 3296967 In the isolated guinea pig ileum, the anticholinergic activity of this drug is about equal to that of atropine; however, when administered orally to unanesthetized cats, it is only about half as active as atropine. In laboratory animals, its antihistaminic activity and duration of action approach those of pyrilamine maleate. INDICATIONS For use as an adjunct in the therapy of all forms of parkinsonism (see DOSAGE AND ADMINISTRATION). Useful also in the control of extrapyramidal disorders (except tardive dyskinesia – see PRECAUTIONS) due to neuroleptic drugs (e.g., phenothiazines). CONTRAINDICATIONS Hypersensitivity to any component of COGENTIN injection. Because of its atropine-like side effects, this drug is contraindicated in pediatric patients under three years of age, and should be used with caution in older pediatric patients. WARNINGS Safe use in pregnancy has not been established. -

Neurochemical and Behavioral Features in Genetic Absence Epilepsy and in Acutely Induced Absence Seizures
Hindawi Publishing Corporation ISRN Neurology Volume 2013, Article ID 875834, 48 pages http://dx.doi.org/10.1155/2013/875834 Review Article Neurochemical and Behavioral Features in Genetic Absence Epilepsy and in Acutely Induced Absence Seizures A. S. Bazyan1 and G. van Luijtelaar2 1 Institute of Higher Nervous Activity and Neurophysiology, Russian Academy of Science, Russian Federation, 5A Butlerov Street, Moscow 117485, Russia 2 Biological Psychology, Donders Centre for Cognition, Donders Institute for Brain, Cognition and Behavior, Radboud University Nijmegen, P.O. Box 9104, 6500 HE Nijmegen, The Netherlands Correspondence should be addressed to G. van Luijtelaar; [email protected] Received 21 January 2013; Accepted 6 February 2013 Academic Editors: R. L. Macdonald, Y. Wang, and E. M. Wassermann Copyright © 2013 A. S. Bazyan and G. van Luijtelaar. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. The absence epilepsy typical electroencephalographic pattern of sharp spikes and slow waves (SWDs) is considered to be dueto an interaction of an initiation site in the cortex and a resonant circuit in the thalamus. The hyperpolarization-activated cyclic nucleotide-gated cationic Ih pacemaker channels (HCN) play an important role in the enhanced cortical excitability. The role of thalamic HCN in SWD occurrence is less clear. Absence epilepsy in the WAG/Rij strain is accompanied by deficiency of the activity of dopaminergic system, which weakens the formation of an emotional positive state, causes depression-like symptoms, and counteracts learning and memory processes. -

Neurophysiological Studies on the Effect of Acetone on Pentylenetetrazole-Induced Seizure in Rats
Bull. Egypt. Soc. Physiol. Sci. 31 (1) 2011 Shehata Neurophysiological Studies on the Effect of Acetone on Pentylenetetrazole-Induced Seizure in Rats Ahmed M. Shehata Department of Physiology- National Organization for Drug Control and Research ABSTRACT Recent interest in the anticonvulsant effects of acetone has stemmed from studies related to the ketogenic diet (KD). Despite knowledge of acetone's anticonvulsant properties, the neurochemical basis for this effect is not well known. The present study aimed to explore the neurochemical basis underlying the anticonvulsant effect of acetone in pentylenetetrazol (PTZ) - induced convulsions. This was achieved through determining the neurochemical changes of acetone in pentylenetetrazol (PTZ) – treated rats. Male adult rats received either saline, acetone (15 m mol/kg i.p.), PTZ (60 mg/kg, i.p.), or acetone 3 h before PTZ injection. Result showed that the maximum concentration of acetone reached about 3 h after acetone administration. Thus, the animals were administered pentylentetrazole three hours after acetone treatment. Pentylenetetrazole treated rats exhibited epilepsy, increased brain levels of excitatory amino acids (glutamate and aspartic acids) and decreased levels of inhibitory amino acid (γ-aminobutyric acid and glycine). In addition, pentylenetetrazole treatment decreased total antioxidant activity and reduced glutathione (GSH) in brain. Acetone pretreatment remarkably decreased seizure incidence rate and increased seizure latency. Moreover, acetone significantly minimized the disturbing effect of pentylenetetrazole on the redox status and the balance between excitatory and inhibitory amino acids. The study indicated that the epilepsy might be mediated, at least partially, through the disturbance in the redox status and imbalance between excitatory and inhibitory amino acids in the brain. -

Pentylenetetrazole Kindling Epilepsy Model Pentilentetrazol Tutuşma Epilepsi Modeli Özlem ERGÜL ERKEÇ, Okan ARIHAN
Epilepsi 2015;21(1):6-12 DOI: 10.5505/epilepsi.2015.08108 REVIEW / DERLEME Pentylenetetrazole Kindling Epilepsy Model Pentilentetrazol Tutuşma Epilepsi Modeli Özlem ERGÜL ERKEÇ, Okan ARIHAN Department of Physiology, Yüzüncü Yıl University Faculty of Medicine, Van Özlem Ergül Erkeç Summary Epilepsy is one of the most common neurologic disorders affecting approximately 1% of the general population, and no alleviation was achieved in one third of the patients medicated with antiepileptic drugs. Current treatments of epilepsy are symptomatic and have more anti-seizure effects than antiepileptic effects. These therapies do not cure epilepsy. Basic mechanisms of epilepsy have not entirely been clarified yet. Using seizure and epilepsy animal models, our comprehension about basic mechanisms underlying epileptogenesis has im- proved. In addition, animal models of epilepsy and seizures are very useful in the discovery and development of new antiepileptic drugs. Pentylenetetrazole (PTZ) is widely used in antiepileptic drug discovery studies, and PTZ kindling model is very important to understand the pathophysiology of epilepsy. In this review, current information about PTZ kindling model was given in this aspect. Key words: Chemical kindling; epilepsy; experimental animal models; pentylenetetrazole; rat. Özet Epilepsi genel popülasyonun yaklaşık %1’ini etkileyen en yaygın nörolojik düzensizliklerden biridir. Antiepileptik ilaç tedavisi gören hastaların üçte birinde hiçbir hafifleme elde edilememektedir. Epilepsi için kullanılan güncel tedaviler semptomatiktir ve antiepileptik etkiden ziyade anti nöbet etkilidir. Bu tedaviler epilepsiyi tedavi etmemektedir. Epilepsinin temel mekanizmaları da henüz tam olarak anlaşılamamıştır. Nö- bet ve epilepsi hayvan modelleri kullanılarak epilepsinin temelinde yatan mekanizmaları kavrayışımız ilerlemektedir. Ayrıca epilepsi ve nöbet hayvan modelleri yeni antiepileptik ilaç keşfi ve gelişimi bakımından kullanışlıdırlar. -

The Timeless Beneficial Bloom Herb Profile Hawthorn Crataegus Monogyna, C
HerbalGram 96 • November 2012 - January 2013 HerbalGram 96 • November 2012 - January 2013 Largest Echinacea Trial • Bilberry Adulteration • Sports and Supplements Federal Cannabis Crackdown • Bacopa and Memory • Butterbur and Migraine The Journal of the American Botanical Council Number 96 | November 2012 - January 2013 Turkish RoseTurkish • Trial Largest • Echinacea Bilberry Adulteration • Sports and Supplements • Cannabis Crackdown Federal • Bacopa and Memory • Butterbur and Migraine US/CAN $6.95 www.herbalgram.org Rose www.herbalgram.org The Timeless Beneficial Bloom Herb Profile Hawthorn Crataegus monogyna, C. laevigata Family: Rosaceae INTRODUCTION HISTORY AND CULTURAL SIGNIFICANCE Hawthorn is a large shrub or small tree (15-30 feet on The generic name, Crataegus, comes from the Greek kratos, average) in the genus Crataegus, native to temperate North meaning hard or strong, referring to the plant’s wood.13,14 America, Europe, and East Asia.1 The plants are indetermi- The common name refers to the plant’s thorns and fruit, nately thorny, with variable shape, and have perfect, radially known as haws, and may also refer to its use to form hedges, symmetrical, 5-petaled white to pink flowers (red in some which were called haws in earlier times.13 Other common M I S S I O N D R I V E N : cultivars) in corymbs (flat-topped clusters).2,3 The red fruit names for C. laevigata include English hawthorn, white are drupes (one-seeded and fleshy) but are commonly called thorn, May tree (referring to when it blooms), and two- berries in the trade. The genus Crataegus comprises approxi- style hawthorn; and English hawthorn, one-seed hawthorn, mately 250-280 species, the most commonly used in West- and one-style hawthorn for C. -

Allosteric Modulation of GABAA Receptors by an Anilino Enaminone in an Olfactory Center of the Mouse Brain
Pharmaceuticals 2014, 7, 1069-1090; doi:10.3390/ph7121069 OPEN ACCESS pharmaceuticals ISSN 1424-8247 www.mdpi.com/journal/pharmaceuticals Review Allosteric Modulation of GABAA Receptors by an Anilino Enaminone in an Olfactory Center of the Mouse Brain Thomas Heinbockel 1,*, Ze-Jun Wang 1 and Patrice L. Jackson-Ayotunde 2 1 Department of Anatomy, College of Medicine, Howard University, Washington, DC 20059, USA; E-Mail: [email protected] 2 Department of Pharmaceutical Sciences, University of Maryland Eastern Shore, Princess Anne, MD 21853, USA; E-Mail: [email protected] * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +1-202-806-9873. External Editor: Marlene A. Jacobson Received: 15 April 2014; in revised form: 24 November 2014 / Accepted: 4 December 2014 / Published: 17 December 2014 Abstract: In an ongoing effort to identify novel drugs that can be used as neurotherapeutic compounds, we have focused on anilino enaminones as potential anticonvulsant agents. Enaminones are organic compounds containing a conjugated system of an amine, an alkene and a ketone. Here, we review the effects of a small library of anilino enaminones on neuronal activity. Our experimental approach employs an olfactory bulb brain slice preparation using whole-cell patch-clamp recording from mitral cells in the main olfactory bulb. The main olfactory bulb is a key integrative center in the olfactory pathway. Mitral cells are the principal output neurons of the main olfactory bulb, receiving olfactory receptor neuron input at their dendrites within glomeruli, and projecting glutamatergic axons through the lateral olfactory tract to the olfactory cortex. The compounds tested are known to be effective in attenuating pentylenetetrazol (PTZ) induced convulsions in rodent models. -
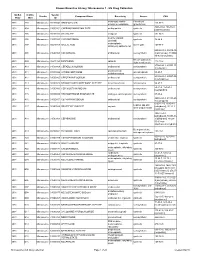
Known Bioactive Library: Microsource 1 - US Drug Collection
Known Bioactive Library: Microsource 1 - US Drug Collection ICCB-L ICCB-L Vendor Vendor Compound Name Bioactivity Source CAS Plate Well ID antifungal, inhibits Penicillium 2091 A03 Microsource 00200046 GRISEOFULVIN 126-07-8 mitosis in metaphase griseofulvum 3505-38-2, 486-16-8 2091 A04 Microsource 01500161 CARBINOXAMINE MALEATE antihistaminic synthetic [carbinoxamine] 2091 A05 Microsource 00200331 SALSALATE analgesic synthetic 552-94-3 muscle relaxant 2091 A06 Microsource 01500162 CARISOPRODOL synthetic 78-44-4 (skeletal) antineoplastic, 2091 A07 Microsource 00210369 GALLIC ACID insect galls 149-91-7 astringent, antibacterial 66592-87-8, 50370-12- 2091 A08 Microsource 01500163 CEFADROXIL antibacterial semisynthetic 2 [anhydrous], 119922- 89-9 [hemihydrate] Rheum palmatum, 2091 A09 Microsource 00211468 DANTHRON cathartic 117-10-2 Xyris semifuscata 27164-46-1, 25953-19- 2091 A10 Microsource 01500164 CEFAZOLIN SODIUM antibacterial semisynthetic 9 [cefazolin] glucocorticoid, 2091 A11 Microsource 00300024 HYDROCORTISONE adrenal glands 50-23-7 antiinflammatory 64485-93-4, 63527-52- 2091 A12 Microsource 01500165 CEFOTAXIME SODIUM antibacterial semisynthetic 6 [cefotaxime] 2091 A13 Microsource 00300029 DESOXYCORTICOSTERONE ACETATE mineralocorticoid adrenocortex 56-47-3 58-71-9, 153-61-7 2091 A14 Microsource 01500166 CEPHALOTHIN SODIUM antibacterial semisynthetic [cephalothin] 2091 A15 Microsource 00300034 TESTOSTERONE PROPIONATE androgen, antineoplastic semisynthetic 57-85-2 24356-60-3, 21593-23- 2091 A16 Microsource 01500167 CEPHAPIRIN SODIUM