Download The
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Questions in the Chemical Enzymology of MAO
Review Questions in the Chemical Enzymology of MAO Rona R. Ramsay 1,* and Alen Albreht 2 1 Biomedical Sciences Research Complex, School of Biology, University of St Andrews, St Andrews KY16 9ST, UK 2 Laboratory for Food Chemistry, Department of Analytical Chemistry, National Institute of Chemistry, Hajdrihova 19, SI-1000 Ljubljana, Slovenia; [email protected] * Correspondence: [email protected]; Tel.: +44-(0)-1334-474740 Abstract: We have structure, a wealth of kinetic data, thousands of chemical ligands and clinical information for the effects of a range of drugs on monoamine oxidase activity in vivo. We have comparative information from various species and mutations on kinetics and effects of inhibition. Nevertheless, there are what seem like simple questions still to be answered. This article presents a brief summary of existing experimental evidence the background and poses questions that remain intriguing for chemists and biochemists researching the chemical enzymology of and drug design for monoamine oxidases (FAD-containing EC 4.1.3.4). Keywords: chemical mechanism; kinetic mechanism; oxidation; protein flexibility; cysteine modifica- tion; reversible/irreversible inhibition; molecular dynamics; simulation 1. Introduction Monoamine oxidase (E.C. 1.4.3.4) enzymes MAO A and MAO B are FAD-containing Citation: Ramsay, R.R.; Albreht, A. proteins located on the outer face of the mitochondrial inner membrane, retained there Questions in the Chemical Enzymology of MAO. Chemistry 2021, by hydrophobic interactions and a transmembrane helix. The redox co-factor (FAD) is 3, 959–978. https://doi.org/10.3390/ covalently attached to a cysteine and buried deep inside the protein [1]. -

Treatment for Calcium Channel Blocker Poisoning a Systematic Review
Clinical Toxicology ISSN: 1556-3650 (Print) 1556-9519 (Online) Journal homepage: http://www.tandfonline.com/loi/ictx20 Treatment for calcium channel blocker poisoning: A systematic review M. St-Onge, P.-A. Dubé, S. Gosselin, C. Guimont, J. Godwin, P. M. Archambault, J.-M. Chauny, A. J. Frenette, M. Darveau, N. Le sage, J. Poitras, J. Provencher, D. N. Juurlink & R. Blais To cite this article: M. St-Onge, P.-A. Dubé, S. Gosselin, C. Guimont, J. Godwin, P. M. Archambault, J.-M. Chauny, A. J. Frenette, M. Darveau, N. Le sage, J. Poitras, J. Provencher, D. N. Juurlink & R. Blais (2014) Treatment for calcium channel blocker poisoning: A systematic review, Clinical Toxicology, 52:9, 926-944, DOI: 10.3109/15563650.2014.965827 To link to this article: http://dx.doi.org/10.3109/15563650.2014.965827 © 2014 The Author(s). Published by Taylor & View supplementary material Francis. Published online: 06 Oct 2014. Submit your article to this journal Article views: 8320 View related articles View Crossmark data Citing articles: 47 View citing articles Full Terms & Conditions of access and use can be found at http://www.tandfonline.com/action/journalInformation?journalCode=ictx20 Download by: [Bird Lib Ouhsc] Date: 14 November 2017, At: 06:23 Clinical Toxicology (2014), 52, 926–944 Copyright © 2014 Informa Healthcare USA, Inc. ISSN: 1556-3650 print / 1556-9519 online DOI: 10.3109/15563650.2014.965827 REVIEW ARTICLE Treatment for calcium channel blocker poisoning: A systematic review M. ST-ONGE,1,2,3 P.-A. DUBÉ,4,5,6 S. GOSSELIN,7,8,9 C. GUIMONT,10 J. -
![Characterization of the Binding and Comparison of the Distribution of Benzodiazepine Receptors Labeled with Ph1diazepam and [3H]Alprazolam L&S K](https://docslib.b-cdn.net/cover/7046/characterization-of-the-binding-and-comparison-of-the-distribution-of-benzodiazepine-receptors-labeled-with-ph1diazepam-and-3h-alprazolam-l-s-k-1187046.webp)
Characterization of the Binding and Comparison of the Distribution of Benzodiazepine Receptors Labeled with Ph1diazepam and [3H]Alprazolam L&S K
fIivIoPSYCHOPHARMACOLOGY 1993-VOL. 8, NO.4 305 Characterization of the Binding and Comparison of the Distribution of Benzodiazepine Receptors Labeled with pH1Diazepam and [3H]Alprazolam l&s K. Wamsley, Ph.D., Lisa L. Longlet, B.S., MaryAnne E. Hunt, Ph.D., Donald R. Mahan, IfiMilrio E. Alburges, Ph.D. Ttrbinding characteristics of [3H]diazepam and benzodiazepine drugs and apparently do not overlap onto �prazolam were obtained by in vitro analysis of other subtypes of receptors. These experiments were IIfiorrsof rat brain. Dissociation, association, and performed by both binding assay in tissue sections and by sDlItionaTUllyses were performed to optimize the light microscopic autoradiography. The major difference GdilWnsfor obtaining selective labeling of between the labeling of the two compounds is· represented llaldiDzepinerecept ors with the two tritiated by the peripheral benzodiazepine sites, which are GIIpOUnds. Both drugs approached equilibrium rapidly recognized by [3H]diazepam, but not occupied by nntro. Rosenthal analysis (Scatchard plot) of the [3H]alprazolam (at nanomolar concentrations). This amiondata indicated a similar finite number of difference was readily apparent in the auto radiograms. IIII;otSwas being occupied by both ligands. Other pharmacokinetic or pharmacodynamic properties Clapetitionstudies, using various ligands to inhibit both must distinguish these two benzodiazepines. fH}bu.tpam and [3H]alprazolam indicated that these [Neuropsychopharmacology 8:305-314, 1993J tIIIcmnpoundsbind to the tissue sections as typical -

Accepted Manuscript
Accepted Manuscript Taurine–magnesium coordination compound, a potential anti- arrhythmic complex, improves aconitine-induced arrhythmias through regulation of multiple ion channels Jianshi Lou, Hong Wu, Lingfang Wang, Lin Zhao, Xin Li, Yi Kang, Ke Wen, Yongqiang Yin PII: S0041-008X(18)30375-2 DOI: doi:10.1016/j.taap.2018.08.008 Reference: YTAAP 14363 To appear in: Toxicology and Applied Pharmacology Received date: 1 March 2018 Revised date: 3 August 2018 Accepted date: 14 August 2018 Please cite this article as: Jianshi Lou, Hong Wu, Lingfang Wang, Lin Zhao, Xin Li, Yi Kang, Ke Wen, Yongqiang Yin , Taurine–magnesium coordination compound, a potential anti-arrhythmic complex, improves aconitine-induced arrhythmias through regulation of multiple ion channels. Ytaap (2018), doi:10.1016/j.taap.2018.08.008 This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain. ACCEPTED MANUSCRIPT Taurine-magnesium Coordination Compound, a Potential Anti-arrhythmic Complex, Improves Aconitine-induced Arrhythmias through Regulation of Multiple Ion Channels Jianshi Lou1, Hong Wu1,2, Lingfang Wang3, Lin Zhao4, Xin Li1, Yi Kang1,Ke Wen1,Yongqiang Yin1* 1 Department of Pharmacology, Tianjin Medical University, Tianjin P. R. China 2 Mudanjiang Medical University, Mudanjiang P. R. China 3 Institute of Translational Medicine, Nanchang University, Nanchang P. -

Testosterone and Cholesterol Vasodilation of Rat Aorta Involves L-Type Calcium Channel Inhibition
Hindawi Publishing Corporation Advances in Pharmacological Sciences Volume 2010, Article ID 534184, 10 pages doi:10.1155/2010/534184 Research Article Testosterone and Cholesterol Vasodilation of Rat Aorta Involves L-Type Calcium Channel Inhibition E. Alvarez,´ 1 E. Cairrao,˜ 1, 2 M. Morgado,1 C. Morais,1 and I. Verde1 1 CICS—Centro de Investigac¸ao˜ em Ciˆencias da Saude,´ Universidade da Beira Interior, Av. Infante D. Henrique, 6200-506 Covilha,˜ Portugal 2 Centro Hospitalar da Cova da Beira E.P.E., Quinta do Alvito, 6200-251 Covilha,˜ Portugal CorrespondenceshouldbeaddressedtoI.Verde,[email protected] Received 8 November 2009; Revised 7 January 2010; Accepted 16 January 2010 Academic Editor: Masahiro Oike Copyright © 2010 E. Alvarez´ et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Testosterone has rapid nongenomic vasodilator effects which could be involved in protective cardiovascular actions. Several authors suggested specific mechanisms to explain this effect, but this matter was not clarified yet. We studied the actions of 2+ testosterone and cholesterol on endothelium-denuded rat aorta and their effects on the L-type Ca current (ICa,L) and potassium current (IK). Testosterone (1–100 μM) totally relaxed, in a rapid and concentration-dependent way, the aortic rings contracted by KCl or by (−)-Bay K8644 (BAY). Cholesterol also fully relaxed the contractions induced by KCl. None of the potassium channel antagonists tested (glibenclamide, tetraethylammonium and 4-aminopyridine) modified significantly the relaxant effect of testosterone. The antagonist of classic testosterone receptors, flutamide, did not modify the vasorelaxant effect of testosterone. -

Calcium Channel Activation: a Different Type of Drug Action (Calcium Flux/Neuroblastoma/Dihydropyridines) STEPHEN B
Proc. Nati. Acad. Sci. USA Vol. 81, pp. 5580-5583, September 1984 Neurobiology Calcium channel activation: A different type of drug action (calcium flux/neuroblastoma/dihydropyridines) STEPHEN B. FREEDMAN* AND RICHARD J. MILLERt Department of Pharmacological and Physiological Sciences, University of Chicago, 947 East 58th Street, Chicago, IL 60637 Communicated by JosefFried, May 14, 1984 ABSTRACT Depolarization of NG108-15 (neuroblasto- plemented medium was replaced every 2 days or when re- ma-glioma) cells causes an increase in 45Ca2+ influx. This ef- quired. fect is blocked by low concentrations of dihydropyridines such 45Ca2+ Uptake Studies. Cells (passage 5-20) were subcul- as nitrendipifie and by other blockers of voltage-sensitive calci- tured onto 60-mm tissue culture plates and induced to differ- um channels such ns D-600, diltiazem, and Cd2 . Two other entiate as above. During the assay, tissue culture plates were dihydropyridines, BAY K8644 and CGP 28392, have the op- supported in an open-air waterbath at 370C. Cells were incu- posite effect. Low concentrations of these compounds enhance bated for 5 min at 37TC before the assay. Details of 45Ca2+ depolarization-induced 45Ca2+ influxes. BAY K8644 is more flux studies are given in the individual figure legends. The effective than CGP 28392. Both agents have no effect on fluxes uptake of 45Ca2+ was measured for increasing periods of measured under nondepolarizing conditions. The effects of time in Hepes (20 mM) buffered Eagle's minimal essential BAY K8644 and CGP 28392 can be inhibited by nitrendipine, medium containing 135.7 mM NaCl, 5 mM KCl, 0.44 mM D-600, diltiazem, or Cd2 . -

Disruption of Cav1.2-Mediated Signaling Is a Pathway for Ketamine-Induced Pathology ✉ Huan Chen1, David H
ARTICLE https://doi.org/10.1038/s41467-020-18167-4 OPEN Disruption of Cav1.2-mediated signaling is a pathway for ketamine-induced pathology ✉ Huan Chen1, David H. Vandorpe1, Xiang Xie1, Seth L. Alper 1, Mark L. Zeidel1 & Weiqun Yu1 The general anesthetic ketamine has been repurposed by physicians as an anti-depressant and by the public as a recreational drug. However, ketamine use can cause extensive pathological changes, including ketamine cystitis. The mechanisms of ketamine’s anti- 1234567890():,; depressant and adverse effects remain poorly understood. Here we present evidence that ketamine is an effective L-type Ca2+ channel (Cav1.2) antagonist that directly inhibits cal- cium influx and smooth muscle contractility, leading to voiding dysfunction. Ketamine pre- vents Cav1.2-mediated induction of immediate early genes and transcription factors, and inactivation of Cav1.2 in smooth muscle mimics the ketamine cystitis phenotype. Our results demonstrate that ketamine inhibition of Cav1.2 signaling is an important pathway mediating ketamine cystitis. In contrast, Cav1.2 agonist Bay k8644 abrogates ketamine-induced smooth muscle dysfunction. Indeed, Cav1.2 activation by Bay k8644 decreases voiding frequency while increasing void volume, indicating Cav1.2 agonists might be effective drugs for treat- ment of bladder dysfunction. 1 Department of Medicine, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA, USA. ✉email: [email protected] NATURE COMMUNICATIONS | (2020) 11:4328 | https://doi.org/10.1038/s41467-020-18167-4 | www.nature.com/naturecommunications 1 ARTICLE NATURE COMMUNICATIONS | https://doi.org/10.1038/s41467-020-18167-4 he anesthetic ketamine has been increasingly used for Results Ttreatment of depression and pain, and increasingly subject Ketamine inhibits BSM contractility. -

Developmental Absence of the O2 Sensitivity of L-Type Calcium
0031-3998/08/6302-0176 PEDIATRIC RESEARCH Vol. 63, No. 2, 2008 Copyright © 2008 International Pediatric Research Foundation, Inc. Printed in U.S.A. Developmental Absence of the O2 Sensitivity of L-Type Calcium Channels in Preterm Ductus Arteriosus Smooth Muscle Cells Impairs O2 Constriction Contributing to Patent Ductus Arteriosus BERNARD THE´ BAUD, XI-CHEN WU, HIDEMI KAJIMOTO, SANDRA BONNET, KYOKO HASHIMOTO, EVANGELOS D. MICHELAKIS, AND STEPHEN L. ARCHER Department of Pediatrics [B.T.], Division of Neonatology, Vascular Biology Group [X.-C.W., H.K., K.H., E.D.M.], University of Alberta, Edmonton, T6G 2J3, AB, Canada; Department of Medicine [S.L.A.], Section of Cardiology, University of Chicago, Chicago, Illinois 60637 ABSTRACT: Patent ductus arteriosus (PDA) complicates the hos- Failure of DA closure after birth complicates the hospital pital course of premature infants. Impaired oxygen (O2)-induced course of preterm infants. PDA is associated with an increased vasoconstriction in preterm ductus arteriosus (DA) contributes to incidence of chronic lung disease, intraventricular hemor- PDA and results, in part, from decreased function/expression of rhage, and necrotizing enterocolitis (4). Both medical and O -sensitive, voltage-gated potassium channels (Kv) in DA smooth 2 surgical interventions to close the DA, although usually ef- muscle cells (DASMCs). This paradigm suggests that activation of fective, are associated with additional morbidity (5). the voltage-sensitive L-type calcium channels (CaL), which increases 2ϩ The crucial role of endothelium-derived relaxing and con- cytosolic calcium ([Ca ]i), is a passive consequence of membrane depolarization. However, effective Kv gene transfer only partially stricting factors in regulating DA tone is well established (6). -

Testosterone and Cholesterol Vasodilation of Rat Aorta Involves L-Type Calcium Channel Inhibition
Hindawi Publishing Corporation Advances in Pharmacological Sciences Volume 2010, Article ID 534184, 10 pages doi:10.1155/2010/534184 Research Article Testosterone and Cholesterol Vasodilation of Rat Aorta Involves L-Type Calcium Channel Inhibition E. Alvarez,´ 1 E. Cairrao,˜ 1, 2 M. Morgado,1 C. Morais,1 and I. Verde1 1 CICS—Centro de Investigac¸ao˜ em Ciˆencias da Saude,´ Universidade da Beira Interior, Av. Infante D. Henrique, 6200-506 Covilha,˜ Portugal 2 Centro Hospitalar da Cova da Beira E.P.E., Quinta do Alvito, 6200-251 Covilha,˜ Portugal CorrespondenceshouldbeaddressedtoI.Verde,[email protected] Received 8 November 2009; Revised 7 January 2010; Accepted 16 January 2010 Academic Editor: Masahiro Oike Copyright © 2010 E. Alvarez´ et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Testosterone has rapid nongenomic vasodilator effects which could be involved in protective cardiovascular actions. Several authors suggested specific mechanisms to explain this effect, but this matter was not clarified yet. We studied the actions of 2+ testosterone and cholesterol on endothelium-denuded rat aorta and their effects on the L-type Ca current (ICa,L) and potassium current (IK). Testosterone (1–100 μM) totally relaxed, in a rapid and concentration-dependent way, the aortic rings contracted by KCl or by (−)-Bay K8644 (BAY). Cholesterol also fully relaxed the contractions induced by KCl. None of the potassium channel antagonists tested (glibenclamide, tetraethylammonium and 4-aminopyridine) modified significantly the relaxant effect of testosterone. The antagonist of classic testosterone receptors, flutamide, did not modify the vasorelaxant effect of testosterone. -
Modulation of Bladder Wall Micromotions Alters Intravesical Pressure Activity in the Isolated Bladder
fphys-09-01937 January 9, 2019 Time: 16:24 # 1 ORIGINAL RESEARCH published: 10 January 2019 doi: 10.3389/fphys.2018.01937 Modulation of Bladder Wall Micromotions Alters Intravesical Pressure Activity in the Isolated Bladder Basu Chakrabarty1†, Dominika A. Bijos2,3†, Bahareh Vahabi4, Francesco Clavica5, Anthony J. Kanai6, Anthony E. Pickering1,2, Christopher H. Fry1 and Marcus J. Drake2,3* 1 School of Physiology, Pharmacology and Neuroscience, Faculty of Life Sciences, University of Bristol, Bristol, United Kingdom, 2 Translational Health Sciences, Bristol Medical School, University of Bristol, Bristol, United Kingdom, 3 Southmead Hospital, Bristol Urological Institute, Bristol, United Kingdom, 4 Department of Applied Sciences, University of West England, Bristol, Bristol, United Kingdom, 5 ARTORG Center for Biomedical Engineering Research, University of Bern, Bern, Switzerland, 6 Department of Medicine, University of Pittsburgh, Pittsburgh, PA, United States Micromotions are phasic contractions of the bladder wall. During urine storage, such phasic activity has little effect on intravesical pressure, however, changed motile activity Edited by: may underlie urodynamic observations such as detrusor overactivity. The potential for Janet R. Keast, The University of Melbourne, Australia bladder motility to affect pressure reflects a summation of the overall movements, Reviewed by: comprising the initiation, propagation, and dissipation components of micromotions. Russ Chess-Williams, In this study, the influence of initiation of micromotions was investigated using calcium Bond University, Australia Anna Serena Nagle, activated chloride channel blocker niflumic acid, and the effect of propagation using Indiana Institute of Technology, blockers of gap junctions. The overall bladder tone was modulated using isoprenaline. United States Isolated tissue strips and whole bladder preparations from juvenile rats were used. -

The Pennsylvania State University the Graduate School College of Medicine L-TYPE CALCIUM CHANNEL REGULATION in the P/Q-TYPE CALC
The Pennsylvania State University The Graduate School College of Medicine L-TYPE CALCIUM CHANNEL REGULATION IN THE P/Q-TYPE CALCIUM CHANNEL MUTANT MOUSE, TOTTERING, A MODEL FOR EPISODIC NEUROLOGICAL DISORDERS A Thesis in Neuroscience by Brandy Ellen Fureman 2001 Brandy Ellen Fureman Submitted in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy August 2001 We approve the thesis of Brandy Ellen Fureman. Date of Signature __________________________________ ________________ Ellen J. Hess Associate Professor of Neuroscience and Anatomy Thesis Advisor Co-Chair of Committee __________________________________ ________________ Robert J. Milner Professor of Neuroscience and Anatomy Director, Graduate Program in Neuroscience Co-Chair of Committee __________________________________ ________________ Melvin L. Billingsley Professor of Pharmacology __________________________________ ________________ Robert Levenson Professor of Pharmacology __________________________________ ________________ Theresa L. Wood Associate Professor of Neuroscience and Anatomy iii ABSTRACT Mutations in calcium channels cause episodic neurological disorders in humans and paroxysmal phenotypes in mice. Tottering mice display stress-induced attacks of a movement disorder due to a P/Q-type calcium channel mutation and cerebellar L-type calcium channel upregulation.L-type calcium channels were assessed in studies of calcium uptake, radioligand binding and in situ hybridization in tottering mice. These studies confirmed the increase in tottering cerebellar L-type calcium channels. Mutant mice also exhibited unique responses to repeated exposure to an L-type calcium channel antagonist (nimodipine) and agonist (BAY K8644). These studies provide further evidence of L-type calcium channel misregulation in the tottering mouse cerebellum. In developing tottering mice, restraint stress did not produce motor attacks until twenty- two days of age; attack frequencies at p22 were similar to adults. -
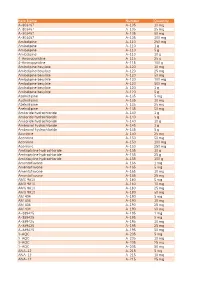
Item Name Catalog Number Quantity A-803467 A-105 10 Mg A
Catalog Item Name Number Quantity A-803467 A-105 10 mg A-803467 A-105 25 mg A-803467 A-105 50 mg A-803467 A-105 100 mg Amlodipine A-110 250 mg Amlodipine A-110 1 g Amlodipine A-110 5 g Amlodipine A-110 10 g 4-Aminopyridine A-115 25 g 4-Aminopyridine A-115 100 g Amlodipine besylate A-120 10 mg Amlodipine besylate A-120 25 mg Amlodipine besylate A-120 50 mg Amlodipine besylate A-120 100 mg Amlodipine besylate A-120 500 mg Amlodipine besylate A-120 1 g Amlodipine besylate A-120 5 g Azelnidipine A-135 5 mg Azelnidipine A-135 10 mg Azelnidipine A-135 25 mg Azelnidipine A-135 50 mg Amiloride hydrochloride A-140 1 g Amiloride hydrochloride A-140 5 g Amiloride hydrochloride A-140 10 g Ambroxol hydrochloride A-145 1 g Ambroxol hydrochloride A-145 5 g Aconitine A-150 25 mg Aconitine A-150 50 mg Aconitine A-150 100 mg Aconitine A-150 250 mg Amitriptyline hydrochloride A-155 10 g Amitriptyline hydrochloride A-155 25 g Amitriptyline hydrochloride A-155 100 g Amentoflavone A-165 1 mg Amentoflavone A-165 5 mg Amentoflavone A-165 10 mg Amentoflavone A-165 25 mg AMG 9810 A-180 5 mg AMG 9810 A-180 10 mg AMG 9810 A-180 25 mg AMG 9810 A-180 50 mg AM 404 A-190 5 mg AM 404 A-190 10 mg AM 404 A-190 25 mg AM 404 A-190 50 mg A-889425 A-195 1 mg A-889425 A-195 5 mg A-889425 A-195 10 mg A-889425 A-195 25 mg A-889425 A-195 50 mg 3-AQC A-205 5 mg 3-AQC A-205 10 mg 3-AQC A-205 25 mg 3-AQC A-205 50 mg ANA-12 A-215 5 mg ANA-12 A-215 10 mg ANA-12 A-215 25 mg ANA-12 A-215 50 mg ANA-12 A-215 100 mg A 967079 A-225 5 mg A 967079 A-225 10 mg A 967079 A-225 25 mg A 967079