Genetic Analysis of Uniparental and Autosomal Markers in Human Populations
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genetic Analysis of the Major Tribes of Buner and Swabi Areas Through Dental Morphology and Dna Analysis
GENETIC ANALYSIS OF THE MAJOR TRIBES OF BUNER AND SWABI AREAS THROUGH DENTAL MORPHOLOGY AND DNA ANALYSIS MUHAMMAD TARIQ DEPARTMENT OF GENETICS HAZARA UNIVERSITY MANSEHRA 2017 I HAZARA UNIVERSITY MANSEHRA Department of Genetics GENETIC ANALYSIS OF THE MAJOR TRIBES OF BUNER AND SWABI AREAS THROUGH DENTAL MORPHOLOGY AND DNA ANALYSIS By Muhammad Tariq This research study has been conducted and reported as partial fulfillment of the requirements of PhD degree in Genetics awarded by Hazara University Mansehra, Pakistan Mansehra The Friday 17, February 2017 I ABSTRACT This dissertation is part of the Higher Education Commission of Pakistan (HEC) funded project, “Enthnogenetic elaboration of KP through Dental Morphology and DNA analysis”. This study focused on five major ethnic groups (Gujars, Jadoons, Syeds, Tanolis, and Yousafzais) of Buner and Swabi Districts, Khyber Pakhtunkhwa Province, Pakistan, through investigations of variations in morphological traits of the permanent tooth crown, and by molecular anthropology based on mitochondrial and Y-chromosome DNA analyses. The frequencies of seven dental traits, of the Arizona State University Dental Anthropology System (ASUDAS) were scored as 17 tooth- trait combinations for each sample, encompassing a total sample size of 688 individuals. These data were compared to data collected in an identical fashion among samples of prehistoric inhabitants of the Indus Valley, southern Central Asia, and west-central peninsular India, as well as to samples of living members of ethnic groups from Abbottabad, Chitral, Haripur, and Mansehra Districts, Khyber Pakhtunkhwa and to samples of living members of ethnic groups residing in Gilgit-Baltistan. Similarities in dental trait frequencies were assessed with C.A.B. -

Genes, Culture, and Agriculture: an Example of Human Niche Construction
Texas A&M University-San Antonio Digital Commons @ Texas A&M University- San Antonio History Faculty Publications College of Arts and Sciences 2012 Genes, Culture, and Agriculture: An Example of Human Niche Construction Michael J. O'Brien Texas A&M University-San Antonio, [email protected] K. N. Laland Follow this and additional works at: https://digitalcommons.tamusa.edu/hist_faculty Part of the Anthropology Commons Repository Citation O'Brien, Michael J. and Laland, K. N., "Genes, Culture, and Agriculture: An Example of Human Niche Construction" (2012). History Faculty Publications. 12. https://digitalcommons.tamusa.edu/hist_faculty/12 This Article is brought to you for free and open access by the College of Arts and Sciences at Digital Commons @ Texas A&M University- San Antonio. It has been accepted for inclusion in History Faculty Publications by an authorized administrator of Digital Commons @ Texas A&M University- San Antonio. For more information, please contact [email protected]. 434 Current Anthropology Volume 53, Number 4, August 2012 Genes, Culture, and Agriculture An Example of Human Niche Construction by Michael J. O’Brien and Kevin N. Laland Theory and empirical data from a variety of disciplines strongly imply that recent human history involves extensive gene-culture coevolution, much of it as a direct result of human agricultural practices. Here we draw on niche- construction theory (NCT) and gene-culture coevolutionary theory (GCT) to propose a broad theoretical framework (NCT-GCT) with which archaeologists and anthropologists can explore coevolutionary dynamics. Humans are enormously potent niche constructors, and understanding how niche construction regulates ecosystem dynamics is central to understanding the impact of human populations on their ecological and developmental environments. -
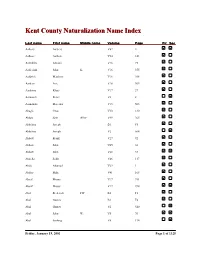
Kent County Naturalization Name Index, Aalbers, A
Kent County Naturalization Name Index Last name First name Middle name Volume Page Fir Sec Aalbers Aalbers V62 4 Aalbers Aalbert V24 141 Aalddriks Antonie V16 75 Aalderink John K. V16 355 Aaldrick Matthew V16 308 Aardem Arie V16 304 Aardema Klaas V17 27 Aarnouds Pieter V6 8 Aarnoudse Marenus V15 503 Abagis Chas V30 130 Abbas Sain Allez V49 265 Abbelma Joseph B1 F5 Abbelma Joseph V2 564 Abbott Frank V27 92 Abbott John V45 36 Abbott John V68 33 Abdella Salik V46 117 Abdo Ahamad V29 1 Abdoo Mike V41 168 Abeaf Moses V17 391 Abeaf Moses V17 394 Abel Frederick FW B1 F1 Abel Gustav B1 F4 Abel Gustav V2 540 Abel John W. V5 70 Abel Ludwig V8 134 Friday, January 19, 2001 Page 1 of 1325 Last name First name Middle name Volume Page Fir Sec Abella Salih V68 85 Abezi Albert V25 76 Aboabsee Theab V74 40 Aboasee Theab V18 150 Abood N. B1 F5 Abood Nemy V3 90 Abraham John V17 381 Abrahamson Charles Y. B7 200 Abram John B1 F1 Abramson Morris B1 F3 Abraursz Abram Peter V27 159 Abromaitis Louis V27 381 Abromaitis Louis V67 90 Absmaier Carl V77 4 Accardi Guiseppe V50 79 Acheson John V16 616 Achille Minciotti V51 142 Achtenhof Jakob V15 145 Achter Jan V17 200 Achterhof Henri B1 F1 Achterhof Johannes V15 500 Achterhof Matheus B1 F1 Achtjes John B7 107 Ackermann Joseph V15 282 Acton John C. B7 222 Adair David G. V15 335 Adair Joseph V15 335 Adalphson Emil V18 197 Friday, January 19, 2001 Page 2 of 1325 Last name First name Middle name Volume Page Fir Sec Adam Frickartz Heinrich V41 279 Adama Jelle V22 176 Adamawiczus Baltris V37 155 Adamczak Peter V38 245 Adamczyk Wladyslaw V35 291 Adams Edward John V24 70 Adams Frank B1 F2 Adams George W. -

History and Geography of Human Y-Chromosome in Europe: a SNP Perspective
JASs Invited Reviews Journal of Anthropological Sciences Vol. 86 (2008), pp. 59-89 History and geography of human Y-chromosome in Europe: a SNP perspective Paolo Francalacci & Daria Sanna Dipartimento di Zoologia e Genetica Evoluzionistica, Università di Sassari, via Muroni 25 – 07100 Sassari, Italy e-mail: [email protected] Summary - e genetic variation observed in the modern European populations can be used to reconstruct the history of the human peopling of the continent. In recent times, a great importance has been given to uniparental markers such as the Y-chromosome. is chromosome, which is passed from father to son, does not have a counterpart subject to recombination and the only possible source of variation is mutation. e nucleotide changes accumulate over time in the molecule, with no rearrangement among lineages. Lately, the D-HPLC technique, which allows the eff ective detection of single nucleotide polymorphisms (SNPs), was used to boost the number of available polymorphisms on the Y-chromosome. Since the year 2000, a number of studies were aimed both at the reconstruction of Y-chromosome phylogeny and the geographic distribution of Y-chromosome variation in Europe. e distribution of distinctive Y-chromosome lineages can also display a correspondence with geography, thus providing patterns of affi nity and clues concerning past human movements. It is therefore possible to recognize the eff ect of the colonization of Europe following the Last Glacial Maximum, both from the western Iberian and the eastern Balkan refuges. Other lineages show a migratory wave from the Near East, consistent with the demic diff usion model of agriculture. -

4 F\Ihli«N%D by the I Llhunntan Catholu* O Ra Ugi Ja Pro** Aoctt'u' 23.H So Ftakl*; A**
? MlOftEST 1. v£# 5721 COTTAGE ^-U1"1- CttlCAGd 57. 1LL. (JRATIS JUM D fc A U O A S t) H A tr O A S L^idSia Ueiuviy Katalike Iffta 1 » Tft. I ItliiumtaJt Daily Vtim4 f\ihli«n%d by the I llhunntan CatholU* O ra ugi Ja Pro** Aoctt'U' 23.H So ftakl*; A**. < Mo**., •. llllnoU 2231 Sn. Oaktry Are., CMcagn t. Kilnote T r teplione — \ Irglnla 7-A04O •41 N Telepttooo — Vlrfrinla 7-«4*0-4| Vienintelis tautinė* Ir tikybine* mlntl»s The tno«t lnflueatlal I.ithuanlan Daily h*imv%ų llenraitla pasaulyje THE LITHUANIAN DAILY, FRIEND tn America No. 232 Kaina 5 oeatal PIRMADIENIS, SPALIO (OCTOBER) 4, 1954 Priee 5 cente VOL. XXXVII • .-.. .. LONDONO KONFERENCIJOJE SUSITARTA i Formoza Peipingo žaidime Šį mėnesį rinksis dar keliose Indijos premjerą Nchru nepaprastai jaudina susišaudymai, vietose nutarimams tvarkyti kurie vyksta Quemoy — Amoy erdvėje tarp abiejų Kinijos vy riausybių karinių pajėgų. Nehru jaudinasi ne be pagrindo, nes jis, turbūt, goriausiai yra painformuotas apie Peipingo nuotai LONDON, spalio 4. — Devynių valstybių konferencija čia kas. baigėsi vakar pilnu susitarimu dėl V. Vokietijos apginklavimo, jai suverenumo grąžinimo ir jos įjungimo į Briuselio ir Atlanto Galop ir žinomi faktai nėra ~ '. ', raminą. Raudonoji Kinija ima Sią Savaitę SlltartlS paktus. telkti prieš Formozą karines Vakar susitarimai pasirašyti viena iš specialių JT organiza- ne kaip formalios sutartys, bet pajėgas (aviaciją ir parašiuti Triesto ginčui baigti . • . ,J... cijų, tačiau žinoma, kad jos yra kaip principų ir jų praktiško . :. ' ,J . ninkus), o JAV laivynas, dabar labai autonomiškosv , o atominės TRIESTE, sp. 4. — Po devy pritaikymo deklaracijos-proto- žymiai sustiprintas, plaukioja energijos organizacija tos auto nių mėnesių derybų Romoje, kolaj. -

Molecular Genealogy of Tusi Lu's Family Reveals Their Paternal
Journal of Human Genetics https://doi.org/10.1038/s10038-019-0618-0 BRIEF COMMUNICATION Molecular genealogy of Tusi Lu’s family reveals their paternal relationship with Jochi, Genghis Khan’s eldest son 1,2 3 1 1,4 1 1 Shao-Qing Wen ● Hong-Bing Yao ● Pan-Xin Du ● Lan-Hai Wei ● Xin-Zhu Tong ● Ling-Xiang Wang ● 1,4 1 5 6 1 7 1 Chuan-Chao Wang ● Bo-Yan Zhou ● Mei-Sen Shi ● Maxat Zhabagin ● Jiucun Wang ● Dan Xu ● Li Jin ● Hui Li1 Received: 9 January 2019 / Revised: 21 April 2019 / Accepted: 6 May 2019 © The Author(s), under exclusive licence to The Japan Society of Human Genetics 2019 Abstract Genghis Khan’s lineage has attracted both academic and general interest because of its mystery and large influence. However, the truth behind the mystery is complicated and continues to confound the scientific study. In this study, we surveyed the molecular genealogy of Northwestern China’s Lu clan who claim to be the descendants of the sixth son of Genghis Khan, Toghan. We also investigated living members of the Huo and Tuo clans, who, according to oral tradition, were close male relatives of Lu clan. Using network analysis, we found that the Y-chromosomal haplotypes of Lu clan 1234567890();,: 1234567890();,: mainly belong to haplogroup C2b1a1b1-F1756, widely prevalent in Altaic-speaking populations, and are closely related to the Tore clan from Kazakhstan, who claim to be the descendants of the first son of Genghis Khan, Jochi. The most recent common ancestor of the special haplotype cluster that includes the Lu clan and Tore clan lived about 1000 years ago (YA), while the Huo and Tuo clans do not share any Y lineages with the Lu clan. -

Darbininkas
boston public LIERARY C -1 F C F L * - - - L -■ * 1 ■ ' :•. i.division COPLEY SQ BOSTON MASS 16 3 tUlUUUUlUUUtlUlltUlUUiiimittiUmilUlllliliia 4--------------------------------------------- 4< I DABARTIES DARBININKAS Lithuanian Semi-Weekly < • :j BILDESIIJOS i Newspaper , Published every , Ua. DARBININKAS TUESDAY and FRIDAY J 366 Broaway, S. Boston 27, Mau. 1 DĖMESIUI! AMERIKOS LIE ' K. ŠVENTO JUOZAPO DARBININKŲ SĄJUNGOS ORGANAS ♦---------------------------------------- • < _________ I Kitą savaitę penktadie- VOL. XXX — No. 76. TEL. SOUth Boston 2680 ANTRADIENIS (Tuesday) SPALIŲ (October) 9 D., 1945 M. TEL. SOUth Boston 2680 FIVE CENTS nio “Darbininko” numeris išeis LDS ir “Darbininko” 3,000 Pagerbė Šv. Pran 30-ties metų gyvavimo su kakties paminėjimui. Ta ciškų Asizietį Rusija Pyksta Ant J. V., Britani me numeryj tilps įvairių straipsnių apie LDS ir I Boston, Mass. — Sekma-; “Darbininką” ir vaizdų. dieni, spalių 7 d. trys tūks jos Dėl Japonijos Ir Rytinės tančiai Trečiojo Ordeno— I Gerb. LDS ir “Darbinin Šv. Pranciškaus AsiziečioI. ko” įkūrėjai, bendradar narių susirinko pagerbti ir! biai, LDS apskričiai ir pagarbinti įžymųjį šven-: kuopos kviečiamos Uitąjįp~ į Šv. Kryžiaus Kated Stalinas Nevažiuosiąs Iš Sovietų Rusi puošti tą “Darbininko” nu rą. merį savo sveikinimais ir J. E. arkivyskupas Ri- jos Į Trijų Didžiųjų Susirinkimą atsiminimais. Visus raš chard J. Cushing, D. D., tus prašome siųsti taip, pasakė turiningą pamoks Londonas, spalių 8 Kaip žinoma, sovietų Ru- kad Jūsų redakcija juos lą apie Šv. Pranciškų, jo Santykiai tarp didžiųjų są-J sijos propagandos mašina gautų spalių 16 d. arba gyvenimą ir darbus. jungininkų — sovietų Ru-! atsargiai skleidė idėją ne prieš tą dieną. Vėliau gau į Kun. Pranciškus Juškai sijos iš vienos pusės ir, tik Rusijoje, bet ir kaimy- ti sveikinimai ir straips tis, Trečiojo Ordeno dvasi- Britanijos ir Jung. -

SAE 2006 World Congress & Exhibition
SAE 2006 World Congress & Exhibition Technical Session Schedule As of 04/09/2006 07:40 pm Monday, April 3 Can Racing Partnerships Change to Accelerate Innovation (Focus on Sports Car Racing) Session Code: ANNUAL108 Room FEV Powertrain Innovation Forum Session Time: 10:00 a.m. Professional Sanctioning Organizations Organizers - Herbert A. Fishel Moderators - Paul F. Pfanner, Founder, Racer Communications Inc. Panelists - James Julow, President, Sports Car Club of America; H. Doug Robinson, Executive Director, International Motor Sports Association; Scott Atherton, President & CEO, American Le Mans Series; Roger Edmondson, President, Grand American Road Racing; Stephane Ratel, CEO, SRO, Ltd. Monday, April 3 Can Racing Partnerships Change to Accelerate Innovation (Focus on Sports Car Racing) Session Code: ANNUAL109 Room FEV Powertrain Innovation Forum Session Time: 2:00 p.m. Automobile Manufacturers Organizers - Herbert A. Fishel Moderators - John McElroy, President, Blue Sky Productions Panelists - Richard T. Brekus, Mgr, Product Planning & Strategy, BMW of North America; John M. Doonan, Mgr, Motorsports Team Dev, Mazda North American Operations; Marcus Haselgrove, Team Manager, Technical Director, B-K Motorsports Inc.; David R. Wilson, TRD Group VP-Strategic Planning, Fin & Ops, Toyota; Steven J. Wesoloski, Road Racing Group Manager, GM Racing, General Motors Corp.; Robert Davis, Senior VP, Quality R&D, Mazda North American Operations; Frank-Steffen Walliser, General Manager Motorsport Strategy, Porsche AG Monday, April 3 Grand Opening Session Code: ANNUAL100 Room AVL Technology Theater (open to all Session Time: 9:30 a.m. Keynote Speakers - Remarks by: Burkhard Goeschel, Member of the Board of Mgmt., BMW AG Develop and Purchasing; Richard O. Schaum, SAE Automotive Vice President; Helmut List, President & CEO, AVL LIST GmbH Monday, April 3 Innovation in the Automotive Industry - An Extended Session Covering Managing Innovation and Driving Innovation with Research Session Code: ANNUAL101 Room AVL Technology Theater (open to all Session Time: 10:00 a.m. -

The Dichotomy Structure of Y Chromosome Haplogroup N
The dichotomy structure of Y chromosome Haplogroup N Kang Hu1☯, Shi Yan2,*☯, Kai Liu4, Chao Ning5, Lan-Hai Wei2, Shi-Lin Li2, Bing Song2, Ge Yu2, Feng Chen1, Li-Jun Liu1, Zhi-Peng Zhao1, Chuan-Chao Wang2, Ya-Jun Yang2, Zhen-Dong Qin2, Jing-Ze Tan2, Fu-Zhong Xue2, Hui Li2, Long-Li Kang1*, Li Jin2,3 1 Key Laboratory of High Altitude Environment and Gene Related to Disease of Tibet Ministry of Education, Tibet University for Nationalities, Xianyang, Shaanxi, China 2 Ministry of Education Key Laboratory of Contemporary Anthropology and Center for Evolutionary Biology, School of Life Sciences and Institutes of Biomedical Sciences, Fudan University, Shanghai 200433, China 3 Chinese Academy of Sciences Key Laboratory of Computational Biology, CAS-MPG Partner Institute for Computational Biology, SIBS, CAS, Shanghai 200031, China 4 Tibet Occupational College of Technology, Lhasa, Tibet 850000, China 5 College of Life Science, Jilin University, Changchun, Jilin 130012, China ☯These authors contribute equally to this article *Correspondence to: S.Y. ([email protected]) or L.L.K. ([email protected]) Running Title: Dichotomy of N Abstract: Haplogroup N-M231 of human Y chromosome is a common clade from Eastern Asia to Northern Europe, being one of the most frequent haplogroups in Altaic and Uralic-speaking populations. Using newly discovered bi-allelic markers from high-throughput DNA sequencing, we largely improved the phylogeny of Haplogroup N, in which 16 subclades could be identified by 33 SNPs. More than 400 males belonging to Haplogroup N in 34 populations in China were successfully genotyped, and populations in Northern Asia and Eastern Europe were also compared together. -

The Enigmatic Origin of Bovine Mtdna Haplogroup R: Sporadic Interbreeding Or an Independent Event of Bos Primigenius Domestication in Italy?
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by PubMed Central The Enigmatic Origin of Bovine mtDNA Haplogroup R: Sporadic Interbreeding or an Independent Event of Bos primigenius Domestication in Italy? Silvia Bonfiglio1, Alessandro Achilli1,2, Anna Olivieri1, Riccardo Negrini3, Licia Colli3, Luigi Liotta4, Paolo Ajmone-Marsan3, Antonio Torroni1, Luca Ferretti1* 1 Dipartimento di Genetica e Microbiologia, Universita` di Pavia, Pavia, Italy, 2 Dipartimento di Biologia Cellulare e Ambientale, Universita` di Perugia, Perugia, Italy, 3 Istituto di Zootecnica, Universita` Cattolica del Sacro Cuore, Piacenza, Italy, 4 Dipartimento di Morfologia, Biochimica, Fisiologia e Produzioni Animali, Universita` di Messina, Messina, Italy Abstract Background: When domestic taurine cattle diffused from the Fertile Crescent, local wild aurochsen (Bos primigenius) were still numerous. Moreover, aurochsen and introduced cattle often coexisted for millennia, thus providing potential conditions not only for spontaneous interbreeding, but also for pastoralists to create secondary domestication centers involving local aurochs populations. Recent mitochondrial genomes analyses revealed that not all modern taurine mtDNAs belong to the shallow macro-haplogroup T of Near Eastern origin, as demonstrated by the detection of three branches (P, Q and R) radiating prior to the T node in the bovine phylogeny. These uncommon haplogroups represent excellent tools to evaluate if sporadic interbreeding or even additional events of cattle domestication occurred. Methodology: The survey of the mitochondrial DNA (mtDNA) control-region variation of 1,747 bovine samples (1,128 new and 619 from previous studies) belonging to 37 European breeds allowed the identification of 16 novel non-T mtDNAs, which after complete genome sequencing were confirmed as members of haplogroups Q and R. -

Facsimile Del Frontespizio Della Tesi Di Dottorato
Alma Mater Studiorum – Università di Bologna DOTTORATO DI RICERCA IN Biodiversità del Evoluzione Ciclo XXVI Settore Concorsuale di afferenza: 05/B1 - Zoologia e Antropologia Settore Scientifico disciplinare: BIO/08 - Antropologia THE GENETIC HISTORY OF ITALIANS: NEW INSIGHTS FROM UNIPARENTALLY-INHERITED MARKERS Presentata da Stefania Sarno Coordinatore Dottorato Relatore Prof.ssa Barbara Mantovani Prof. Davide Pettener Esame Finale – Anno 2014 Table of contents Table of contents GENERAL INTRODUCTION AND THESIS RATIONAL 1 1. Reconstructing human evolutionary history through genetic tools 2 2. A multi-level analysis approach in the study of the Italian population history and 3 genetic structure: from macro- to micro-geographic perspectives 3. Work outline 5 PART 1 - The genetic history and population structure of Italy: 11 a macro-geographic overview from the uniparental markers 1.1 Introduction 13 1.1.1 Pre-historical and historical population dynamics 14 1.1.1.1 Palaeolithic occupation and post-glacial re-expansions 14 1.1.1.2 Main migration patterns associated to the Neolithic transition 20 1.1.1.3 Population dynamics in historical times 29 1.1.2 A genetic overview on the Italian variability 34 1.1.2.1 Classical genetic markers 34 1.1.2.2 Uniparental molecular markers 38 1.1.2.3 Genome-wide based studies 42 1.2 Specific aims of the studies 45 1.3 Results and discussion 49 Article 1: 51 Boattini A, Martinez-Cruz B, Sarno S, Harmant C, Useli A, Sanz P, Yang-Yao D, Manry J, Ciani G, Luiselli D, Quintana-Murci L, Comas D, Pettener D, Genographic Consortium (2013) Uniparental markers in Italy reveal a sex-biased genetic structure and different historical strata. -

Association Between Mitochondrial DNA Haplogroup Variation and Autism Spectrum Disorders
Research JAMA Psychiatry | Original Investigation Association Between Mitochondrial DNA Haplogroup Variation and Autism Spectrum Disorders Dimitra Chalkia, PhD; Larry N. Singh, PhD; Jeremy Leipzig, MS; Maria Lvova, MD; Olga Derbeneva, PhD; Anita Lakatos, PhD; Dexter Hadley, MD, PhD; Hakon Hakonarson, MD, PhD; Douglas C. Wallace, PhD Supplemental content IMPORTANCE Autism spectrum disorders (ASD) are characterized by impairments in social interaction, communication, and repetitive or restrictive behavior. Although multiple physiologic and biochemical studies have reported defects in mitochondrial oxidative phosphorylation in patients with ASD, the role of mitochondrial DNA (mtDNA) variation has remained relatively unexplored. OBJECTIVE To assess what impact mitochondrial lineages encompassing ancient mtDNA functional polymorphisms, termed haplogroups, have on ASD risk. DESIGN, SETTING, AND PARTICIPANTS In this cohort study, individuals with autism and their families were studied using the Autism Genetic Resource Exchange cohort genome-wide association studies data previously generated at the Children’s Hospital of Philadelphia. From October 2010 to January 2017, we analyzed the data and used the mtDNA single-nucleotide polymorphisms interrogated by the Illumina HumanHap 550 chip to determine the mtDNA haplogroups of the individuals. Taking into account the familial structure of the Autism Genetic Resource Exchange data, we then determined whether the mtDNA haplogroups correlate with ASD risk. MAIN OUTCOMES AND MEASURES Odds ratios of mitochondrial haplogroup as predictors of ASD risk. RESULTS Of 1624 patients with autism included in this study, 1299 were boys (80%) and 325 were girls (20%). Families in the Autism Genetic Resource Exchange collection (933 families, encompassing 4041 individuals: 1624 patients with ASD and 2417 healthy parents and siblings) had been previously recruited in the United States with no restrictions on age, sex, race/ethnicity, or socioeconomic status.