Biocontainers Documentation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Color Chap 2.Cdr
Chapter 2 - Location and coordinates Updated 10 July 2006 -4 -3 -2 -1 0 +1 +2 +3 +4 Figure 2.1 A simple number line acts as a one-dimensional coordinate system. Each number describes the distance of a particular point-location from the origin, or zero. The unit of measure for the distance is arbitrary. 10 9 8 (4.5,7.5) 7 6 s i x a (3,5) - 5 y 4 3 (6,3) 2 1 0 0 1 2 3 4 5 6 7 8 9 10 x - axis Figure 2.2 Here is a simple x-y coordinate system with the coordinates of a few points shown as examples. Coordinates are given as an ordered pair of numbers, with the x-coordinate first. The origin is the lower left, at (0,0). The reference lines are the x and y axes. All grid lines are drawn parallel to the two reference lines. The purpose of the grid is to make it easier to make distance measurements between a location-point and the reference lines. 5 4 3 (2,3) 2 s 1 i x a (-2,0) - 0 y -1 -2 (1,-2) -3 -4 -5 -5 -4 -3 -2 -1 0 1 2 3 4 5 x - axis Figure 2.3 A more general coordinate grid places the origin (0,0) at the center of the grid. The coordinates may have either positive or negative values. The sign merely indicates whether the point is left or right (x), or above or below (y) the axis. -

Educator's Guide: Orion
Legends of the Night Sky Orion Educator’s Guide Grades K - 8 Written By: Dr. Phil Wymer, Ph.D. & Art Klinger Legends of the Night Sky: Orion Educator’s Guide Table of Contents Introduction………………………………………………………………....3 Constellations; General Overview……………………………………..4 Orion…………………………………………………………………………..22 Scorpius……………………………………………………………………….36 Canis Major…………………………………………………………………..45 Canis Minor…………………………………………………………………..52 Lesson Plans………………………………………………………………….56 Coloring Book…………………………………………………………………….….57 Hand Angles……………………………………………………………………….…64 Constellation Research..…………………………………………………….……71 When and Where to View Orion…………………………………….……..…77 Angles For Locating Orion..…………………………………………...……….78 Overhead Projector Punch Out of Orion……………………………………82 Where on Earth is: Thrace, Lemnos, and Crete?.............................83 Appendix………………………………………………………………………86 Copyright©2003, Audio Visual Imagineering, Inc. 2 Legends of the Night Sky: Orion Educator’s Guide Introduction It is our belief that “Legends of the Night sky: Orion” is the best multi-grade (K – 8), multi-disciplinary education package on the market today. It consists of a humorous 24-minute show and educator’s package. The Orion Educator’s Guide is designed for Planetarians, Teachers, and parents. The information is researched, organized, and laid out so that the educator need not spend hours coming up with lesson plans or labs. This has already been accomplished by certified educators. The guide is written to alleviate the fear of space and the night sky (that many elementary and middle school teachers have) when it comes to that section of the science lesson plan. It is an excellent tool that allows the parents to be a part of the learning experience. The guide is devised in such a way that there are plenty of visuals to assist the educator and student in finding the Winter constellations. -

Sydney Observatory Night Sky Map September 2012 a Map for Each Month of the Year, to Help You Learn About the Night Sky
Sydney Observatory night sky map September 2012 A map for each month of the year, to help you learn about the night sky www.sydneyobservatory.com This star chart shows the stars and constellations visible in the night sky for Sydney, Melbourne, Brisbane, Canberra, Hobart, Adelaide and Perth for September 2012 at about 7:30 pm (local standard time). For Darwin and similar locations the chart will still apply, but some stars will be lost off the southern edge while extra stars will be visible to the north. Stars down to a brightness or magnitude limit of 4.5 are shown. To use this chart, rotate it so that the direction you are facing (north, south, east or west) is shown at the bottom. The centre of the chart represents the point directly above your head, called the zenith, and the outer circular edge represents the horizon. h t r No Star brightness Moon phase Last quarter: 08th Zero or brighter New Moon: 16th 1st magnitude LACERTA nd Deneb First quarter: 23rd 2 CYGNUS Full Moon: 30th rd N 3 E LYRA th Vega W 4 LYRA N CORONA BOREALIS HERCULES BOOTES VULPECULA SAGITTA PEGASUS DELPHINUS Arcturus Altair EQUULEUS SERPENS AQUILA OPHIUCHUS SCUTUM PISCES Moon on 23rd SERPENS Zubeneschamali AQUARIUS CAPRICORNUS E SAGITTARIUS LIBRA a Saturn Centre of the Galaxy Antares Zubenelgenubi t s Antares VIRGO s t SAGITTARIUS P SCORPIUS P e PISCESMICROSCOPIUM AUSTRINUS SCORPIUS Mars Spica W PISCIS AUSTRINUS CORONA AUSTRALIS Fomalhaut Centre of the Galaxy TELESCOPIUM LUPUS ARA GRUSGRUS INDUS NORMA CORVUS INDUS CETUS SCULPTOR PAVO CIRCINUS CENTAURUS TRIANGULUM -

THE CONSTELLATION MUSCA, the FLY Musca Australis (Latin: Southern Fly) Is a Small Constellation in the Deep Southern Sky
THE CONSTELLATION MUSCA, THE FLY Musca Australis (Latin: Southern Fly) is a small constellation in the deep southern sky. It was one of twelve constellations created by Petrus Plancius from the observations of Pieter Dirkszoon Keyser and Frederick de Houtman and it first appeared on a 35-cm diameter celestial globe published in 1597 in Amsterdam by Plancius and Jodocus Hondius. The first depiction of this constellation in a celestial atlas was in Johann Bayer's Uranometria of 1603. It was also known as Apis (Latin: bee) for two hundred years. Musca remains below the horizon for most Northern Hemisphere observers. Also known as the Southern or Indian Fly, the French Mouche Australe ou Indienne, the German Südliche Fliege, and the Italian Mosca Australe, it lies partly in the Milky Way, south of Crux and east of the Chamaeleon. De Houtman included it in his southern star catalogue in 1598 under the Dutch name De Vlieghe, ‘The Fly’ This title generally is supposed to have been substituted by La Caille, about 1752, for Bayer's Apis, the Bee; but Halley, in 1679, had called it Musca Apis; and even previous to him, Riccioli catalogued it as Apis seu Musca. Even in our day the idea of a Bee prevails, for Stieler's Planisphere of 1872 has Biene, and an alternative title in France is Abeille. When the Northern Fly was merged with Aries by the International Astronomical Union (IAU) in 1929, Musca Australis was given its modern shortened name Musca. It is the only official constellation depicting an insect. Julius Schiller, who redrew and named all the 88 constellations united Musca with the Bird of Paradise and the Chamaeleon as mother Eve. -

Astrometric Radial Velocities
A&A 381, 446–463 (2002) Astronomy DOI: 10.1051/0004-6361:20011458 & c ESO 2002 Astrophysics Astrometric radial velocities III. Hipparcos measurements of nearby star clusters and associations?,?? Søren Madsen, Dainis Dravins, and Lennart Lindegren Lund Observatory, Box 43, 22100 Lund, Sweden e-mail: soren, dainis, [email protected] Received 10 September 2001 / Accepted 15 October 2001 Abstract. Radial motions of stars in nearby moving clusters are determined from accurate proper motions and trigonometric parallaxes, without any use of spectroscopy. Assuming that cluster members share the same ve- locity vector (apart from a random dispersion), we apply a maximum-likelihood method on astrometric data from Hipparcos to compute radial and space velocities (and their dispersions) in the Ursa Major, Hyades, Coma Berenices, Pleiades, and Praesepe clusters, and for the Scorpius-Centaurus, α Persei, and “HIP 98321” associa- tions. The radial motion of the Hyades cluster is determined to within 0.4 km s−1 (standard error), and that of its individual stars to within 0.6 km s−1. For other clusters, Hipparcos data yield astrometric radial velocities with typical accuracies of a few km s−1. A comparison of these astrometric values with spectroscopic radial ve- locities in the literature shows a good general agreement and, in the case of the best-determined Hyades cluster, also permits searches for subtle astrophysical differences, such as evidence for enhanced convective blueshifts of F-dwarf spectra, and decreased gravitational redshifts in giants. Similar comparisons for the Scorpius OB2 com- plex indicate some expansion of its associations, albeit slower than expected from their ages. -

Debian \ Amber \ Arco-Debian \ Arc-Live \ Aslinux \ Beatrix
Debian \ Amber \ Arco-Debian \ Arc-Live \ ASLinux \ BeatriX \ BlackRhino \ BlankON \ Bluewall \ BOSS \ Canaima \ Clonezilla Live \ Conducit \ Corel \ Xandros \ DeadCD \ Olive \ DeMuDi \ \ 64Studio (64 Studio) \ DoudouLinux \ DRBL \ Elive \ Epidemic \ Estrella Roja \ Euronode \ GALPon MiniNo \ Gibraltar \ GNUGuitarINUX \ gnuLiNex \ \ Lihuen \ grml \ Guadalinex \ Impi \ Inquisitor \ Linux Mint Debian \ LliureX \ K-DEMar \ kademar \ Knoppix \ \ B2D \ \ Bioknoppix \ \ Damn Small Linux \ \ \ Hikarunix \ \ \ DSL-N \ \ \ Damn Vulnerable Linux \ \ Danix \ \ Feather \ \ INSERT \ \ Joatha \ \ Kaella \ \ Kanotix \ \ \ Auditor Security Linux \ \ \ Backtrack \ \ \ Parsix \ \ Kurumin \ \ \ Dizinha \ \ \ \ NeoDizinha \ \ \ \ Patinho Faminto \ \ \ Kalango \ \ \ Poseidon \ \ MAX \ \ Medialinux \ \ Mediainlinux \ \ ArtistX \ \ Morphix \ \ \ Aquamorph \ \ \ Dreamlinux \ \ \ Hiwix \ \ \ Hiweed \ \ \ \ Deepin \ \ \ ZoneCD \ \ Musix \ \ ParallelKnoppix \ \ Quantian \ \ Shabdix \ \ Symphony OS \ \ Whoppix \ \ WHAX \ LEAF \ Libranet \ Librassoc \ Lindows \ Linspire \ \ Freespire \ Liquid Lemur \ Matriux \ MEPIS \ SimplyMEPIS \ \ antiX \ \ \ Swift \ Metamorphose \ miniwoody \ Bonzai \ MoLinux \ \ Tirwal \ NepaLinux \ Nova \ Omoikane (Arma) \ OpenMediaVault \ OS2005 \ Maemo \ Meego Harmattan \ PelicanHPC \ Progeny \ Progress \ Proxmox \ PureOS \ Red Ribbon \ Resulinux \ Rxart \ SalineOS \ Semplice \ sidux \ aptosid \ \ siduction \ Skolelinux \ Snowlinux \ srvRX live \ Storm \ Tails \ ThinClientOS \ Trisquel \ Tuquito \ Ubuntu \ \ A/V \ \ AV \ \ Airinux \ \ Arabian -

Sydney Observatory Night Sky Map December 2014 a Map for Each Month of the Year, to Help You Learn About the Night Sky
Sydney Observatory night sky map December 2014 A map for each month of the year, to help you learn about the night sky www.sydneyobservatory.com.au This star chart shows the stars and constellations visible in the night sky for Sydney, Melbourne, Brisbane, Canberra, Hobart, Adelaide and Perth for December 2014 at about 7:30 pm (Local Standard Time) or 8:30 pm (Local Daylight Savings Time). For Darwin and similar locations the chart will still apply, but some stars will be lost off the southern edge while extra stars will be visible to the north. Stars down to a brightness or magnitude limit of 4.5 are shown. To use this star chart, rotate it so that the direction you are facing (north, south, east or west) is shown at the bottom. The centre of the chart represents the point directly above your head, called the zenith, and the outer circular edge represents the horizon. h t r o N Star Brightness Moon Phase Full Moon: 06th Zero or brighterCapella st Last quarter: 14th 1 AURIGA magnitude Deneb New Moon: 22nd nd LACERTA 2 PERSEUS CYGNUSFirst quarter: 29th rd N Andromeda Galaxy 3 E th ANDROMEDA W 4 N M45 (Pleiades or Seven Sisters) TRIANGULUM Hamal ARIES VULPECULA PEGASUS TAURUS TAURUS Aldebaran AldebaranHyades PISCES Moon on 29th DELPHINUS SAGITTA First point of Aries EQUULEUS Mira Altair Betelgeuse Betelgeuse ORION CETUS ORION AQUARIUS Orion's belt M42 ERIDANUS E Rigel AQUILA a t s Saucepan SCULPTOR Fomalhaut CAPRICORNUS s t FORNAX Moon on 23rd PISCIS AUSTRINUS e PISCIS AUSTRINUS Mars P LEPUS W PHOENIX GRUS MICROSCOPIUM ERIDANUS SCUTUM Sirius -

The National Optical Astronomy Observatory's Dark Skies and Energy Education Program Constellation at Your Fingertips: Crux 1
The National Optical Astronomy Observatory’s Dark Skies and Energy Education Program Constellation at Your Fingertips: Crux Grades: 3rd – 8th grade Overview: Constellation at Your Fingertips introduces the novice constellation hunter to a method for spotting the main stars in the constellation Crux, the Cross. Students will make an outline of the constellation used to locate the stars in Crux. This activity will engage children and first-time night sky viewers in observations of the night sky. The lesson links history, literature, and astronomy. The simplicity of Crux makes learning to locate a constellation and observing exciting for young learners. All materials for Globe at Night are available at http://www.globeatnight.org Purpose: Students will look for the faintest stars visible and record that data in order to compare data in Globe at Night across the world. In many cases, multiple night observations will build knowledge of how the “limiting” stellar magnitudes for a location change overtime. Why is this important to astronomers? Why do we see more stars in some locations and not others? How does this change over time? The focus is on light pollution and the options we have as consumers when purchasing outdoor lighting. The impact in our environment is an important issue in a child’s world. Crux is a good constellation to observe with young children. The constellation Crux (also known as the Southern Cross) is easily visible from the southern hemisphere at practically any time of year. For locations south of 34°S, Crux is circumpolar and thus always visible in the night sky. -
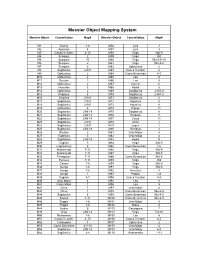
Messier Object Mapping System
Messier Object Mapping System Messier Object Constellation Map# Messier Object Constellation Map# M1 Taurus 1-6 M56 Lyra 3 M2 Aquarius 8 M57 Lyra 3 M3 Canes Venatici 4-10 M58 Virgo IN2-5 M4 Scorpius 2 M59 Virgo IN2 M5 Scorpius 10 M60 Virgo IN2-5-9-10 M6 Scorpius 2 M61 Virgo IN2-5-8 M7 Scorpius 2 M62 Ophiuchus 2 M8 Sagittarius 2-IN1 M63 Canes Venatici 4 M9 Ophiuchus 2 M64 Coma Berenices 4-5 M10 Ophiuchus 2 M65 Leo 5 M11 Scutum 2 M66 Leo 5 M12 Ophiuchus 2 M67 Cancer 6 M13 Hercules 3 M68 Hydra 9 M14 Ophiuchus 2 M69 Sagittarius 2-IN1-8 M15 Pegasus 8 M70 Sagittarius 2-IN1-8 M16 Serpens 2-IN1 M71 Sagittarius 2 M17 Sagittarius 2-IN1 M72 Aquarius 8 M18 Sagittarius 2-IN1 M73 Aquarius 8 M19 Ophiuchus 2 M74 Pisces 11 M20 Sagittarius 2-IN1-8 M75 Sagittarius 8 M21 Sagittarius 2-IN1-8 M76 Perseus 7 M22 Sagittarius 2-IN1-8 M77 Cetus 11 M23 Sagittarius 2-IN1 M78 Orion 1 M24 Sagittarius 2-IN1 M79 Lepus 1 M25 Sagittarius 2-IN1-8 M80 Scorpius 2 M26 Scutum 2 M81 Ursa Major 4 M27 Vupecula 3 M82 Ursa Major 4 M28 Sagittarius 2-IN1-8 M83 Hydra 9 M29 Cygnus 3 M84 Virgo IN2-5 M30 Capricornus 8 M85 Coma Berenices 4-5 M31 Andromeda 7-11 M86 Virgo IN2-5 M32 Andromeda 7-11 M87 Virgo IN2-5 M33 Triangulum 7-11 M88 Coma Berenices IN2-5 M34 Perseus 7-11 M89 Virgo IN2 M35 Gemini 1-6 M90 Virgo IN2-5 M36 Auriga 1-6 M91 Virgo IN2-5 M37 Auriga 1-6 M92 Hercules 3 M38 Auriga 1 M93 Puppis 1-6 M39 Cygnus 3-7 M94 Canes Venatici 4-5 M40 Ursa Major 4 M95 Leo 5 M41 Canis Major 1 M96 Leo 5 M42 Orion 1 M97 Ursa Major 4 M43 Orion 1 M98 Coma Berenices IN2-4-5 M44 Sagittarius 6 M99 Coma Berenices IN2-4-5 M45 Taurus 1-11 M100 Coma Berenices IN2-4-5 M46 Puppis 1-6 M101 Ursa Major 4 M47 Puppis 1-6 M102 Draco 4 M48 Hydra 6 M103 Cassiopeia 7 M49 Virgo 2-IN1-8 M104 Virgo 5-9-10 M50 Monoceros 1-6 M105 Leo 5 M51 Canes Venatici 4-10 M106 Canes Venatici 4-5 M52 Cassiopeia 7 M107 Ophiuchus 2 M53 Coma Berenices 4-5-10 M108 Ursa Major 4 M54 Sagittarius 2-IN1-8 M109 Ursa Major 4-5 M55 Sagittarius 2-8 M110 Andromeda 7-11 Messier Object List # NGC# Constellation Type Name, If Any Mag. -

THE SKY TONIGHT Constellation Is Said to Represent Ganymede, the Handsome Prince of Capricornus Troy
- October Oketopa HIGHLIGHTS Aquarius and Aquila In Greek mythology, the Aquarius THE SKY TONIGHT constellation is said to represent Ganymede, the handsome prince of Capricornus Troy. His good looks attracted the attention of Zeus, who sent the eagle The Greeks associated Capricornus Aquila to kidnap him and carry him with Aegipan, who was one of the to Olympus to serve as a cupbearer Panes - a group of half-goat men to the gods. Because of this story, who often had goat legs and horns. Ganymede was sometimes seen as the god of homosexual relations. He Aegipan assumed the form of a fish- also gives his name to one of the tailed goat and fled into the ocean moons of Jupiter, which are named to flee the great monster Typhon. after the lovers of Zeus. Later, he aided Zeus in defeating Typhon and was rewarded by being To locate Aquarius, first find Altair, placed in the stars. the brightest star in the Aquila constellation. Altair is one of the To find Capricornus (highlighted in closest stars to Earth that can be seen orange on the star chart), first locate with the naked eye, at a distance the Aquarius constellation, then of 17 light years. From Altair, scan look to the south-west along the east-south-east to find Aquarius ecliptic line (the dotted line on the (highlighted in yellow on the star chart). star chart). What’s On in October? October shows at Perpetual Guardian Planetarium, book at Museum Shop or online. See website for show times and - details: otagomuseum.nz October Oketopa SKY GUIDE Capturing the Cosmos Planetarium show. -

School of Arts and Humanities
STUDENT WARNING: This course syllabus is from a previous semester archive and serves only as a preparatory reference. Please use this syllabus as a reference only until the professor opens the classroom and you have access to the updated course syllabus. Please do NOT purchase any books or start any work based on this syllabus; this syllabus may NOT be the one that your individual instructor uses for a course that has not yet started. If you need to verify course textbooks, please refer to the online course description through your student portal. This syllabus is proprietary material of APUS. School of Arts and Humanities MILH 552 The Second World War in the Pacific 3 Credit Hours Length of Course – 8 Weeks Graduate students are encouraged to take required or core courses prior to enrolling in the seminars, concentration courses or electives. Table of Contents Instructor Information Course Materials Course Description Evaluation Procedures Course Scope Course Outline Course Objectives Online Research Services Course Delivery Method Selected Bibliography Instructor Information Table of Contents Course Description This course is a comprehensive analysis of the fighting during World War II in the China-Burma-India Theater, New Guinea, the Solomon Islands, the Aleutians, and the Philippines. Students assess the amphibious campaigns in the central Pacific theatre and evaluate the impact of one of the most massive movements of men and arms in history. Primary focus is on land warfare, its strategies, tactics, leaders and lessons learned. Table of Contents STUDENT WARNING: This course syllabus is from a previous semester archive and serves only as a preparatory reference. -

Nix(OS) - Revolutionizing Packaging and Configuration Management!
Nix(OS) - Revolutionizing packaging and configuration management! The Purely Functional Linux Distribution 1 Before we begin (FYI) Ask questions at any time Please ask lots of questions :) The slides contain some redundancy There are a few optional slides at the end Please give me feedback Louder Faster/slower More/less details Etc. 2 About me Michael Weiss aka. primeos Computer science student at the University of Tübingen I love free soware, etc. First nixpkgs commit: 2016-10-05 I maintain ~41 packages and ~3 modules (2018-06-08) I also love privacy (i.e. no more details :P) Email: [email protected] (#privacy) 3 Main components Nix (package manager) Nixpkgs (Nix packages collection) NixOS (operating system) NixOps (DevOps / cloud deployment tool) 4 Nix* ISO/OSI model NixOps NixOS Nixpkgs Nix 5 Other tools Hydra (Nix based continuous build system) Disnix (distributed services deployment) PatchELF (change dynamic linker and RPATH) {cabal,go,node,pip,python,pypi,composer,hex,bower,vim,...}2 6 History Started as a research project (with funding) First paper in 2004 (many will follow) Nix package manager developed by Eelco Dolstra as part of his PhD research (~2003) First NixOS prototype developed by Armijn Hemel as his master's thesis project Hydra developed as part of the LaQuSo Buildfarm project 7 Timeline 2003: init (research begins) 2007: NixOS becomes usable + x86_64 support 2008: Website moved to nixos.org 2009: Nix logo + Nix(OS) build on Hydra 2011: Migration from Subversion to Git(Hub) 2013: Switch from Upstart to systemd +