Large-Scale Discovery of ERK2 Substrates Identifies ERK-Mediated Transcriptional Regulation by ETV3
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Regulation of Cdc42 and Its Effectors in Epithelial Morphogenesis Franck Pichaud1,2,*, Rhian F
© 2019. Published by The Company of Biologists Ltd | Journal of Cell Science (2019) 132, jcs217869. doi:10.1242/jcs.217869 REVIEW SUBJECT COLLECTION: ADHESION Regulation of Cdc42 and its effectors in epithelial morphogenesis Franck Pichaud1,2,*, Rhian F. Walther1 and Francisca Nunes de Almeida1 ABSTRACT An overview of Cdc42 Cdc42 – a member of the small Rho GTPase family – regulates cell Cdc42 was discovered in yeast and belongs to a large family of small – polarity across organisms from yeast to humans. It is an essential (20 30 kDa) GTP-binding proteins (Adams et al., 1990; Johnson regulator of polarized morphogenesis in epithelial cells, through and Pringle, 1990). It is part of the Ras-homologous Rho subfamily coordination of apical membrane morphogenesis, lumen formation and of GTPases, of which there are 20 members in humans, including junction maturation. In parallel, work in yeast and Caenorhabditis elegans the RhoA and Rac GTPases, (Hall, 2012). Rho, Rac and Cdc42 has provided important clues as to how this molecular switch can homologues are found in all eukaryotes, except for plants, which do generate and regulate polarity through localized activation or inhibition, not have a clear homologue for Cdc42. Together, the function of and cytoskeleton regulation. Recent studies have revealed how Rho GTPases influences most, if not all, cellular processes. important and complex these regulations can be during epithelial In the early 1990s, seminal work from Alan Hall and his morphogenesis. This complexity is mirrored by the fact that Cdc42 can collaborators identified Rho, Rac and Cdc42 as main regulators of exert its function through many effector proteins. -

The Borg Family of Cdc42 Effector Proteins Cdc42ep1–5
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Institute of Cancer Research Repository Biochemical Society Transactions (2016) 0 1–8 DOI: 10.1042/BST20160219 1 2 The Borg family of Cdc42 effector proteins 3 4 Cdc42EP1–5 5 6 Aaron J. Farrugia and Fernando Calvo 7 8 Tumour Microenvironment Team, Division of Cancer Biology, Institute of Cancer Research, 237 Fulham Road, London SW2 6JB, U.K. 9 Correspondence: Fernando Calvo ([email protected]) 10 11 12 13 Despite being discovered more than 15 years ago, the Borg (binder of Rho GTPases) 14 – family of Cdc42 effector proteins (Cdc42EP1 5) remains largely uncharacterised and rela- 15 tively little is known about their structure, regulation and role in development and disease. 16 Recent studies are starting to unravel some of the key functional and mechanistic 17 aspects of the Borg proteins, including their role in cytoskeletal remodelling and signal- 18 ling. In addition, the participation of Borg proteins in important cellular processes such as 19 cell shape, directed migration and differentiation is slowly emerging, directly linking Borgs 20 with important physiological and pathological processes such as angiogenesis, neuro- 21 fi transmission and cancer-associated desmoplasia. Here, we review some of these nd- 22 ings and discuss future prospects. 23 24 25 26 27 28 29 Introduction 30 The Rho GTPase family member Cdc42 regulates a diverse range of cellular functions including cyto- 31 kinesis, cytoskeletal remodelling and cell polarity [1,2]. Like other Rho family members, Cdc42 cycles 32 between two tightly regulated conformational states, a GTP-bound active state and a GDP-bound 33 inactive state [3]. -

Transcriptome Sequencing and Genome-Wide Association Analyses Reveal Lysosomal Function and Actin Cytoskeleton Remodeling in Schizophrenia and Bipolar Disorder
Molecular Psychiatry (2015) 20, 563–572 © 2015 Macmillan Publishers Limited All rights reserved 1359-4184/15 www.nature.com/mp ORIGINAL ARTICLE Transcriptome sequencing and genome-wide association analyses reveal lysosomal function and actin cytoskeleton remodeling in schizophrenia and bipolar disorder Z Zhao1,6,JXu2,6, J Chen3,6, S Kim4, M Reimers3, S-A Bacanu3,HYu1, C Liu5, J Sun1, Q Wang1, P Jia1,FXu2, Y Zhang2, KS Kendler3, Z Peng2 and X Chen3 Schizophrenia (SCZ) and bipolar disorder (BPD) are severe mental disorders with high heritability. Clinicians have long noticed the similarities of clinic symptoms between these disorders. In recent years, accumulating evidence indicates some shared genetic liabilities. However, what is shared remains elusive. In this study, we conducted whole transcriptome analysis of post-mortem brain tissues (cingulate cortex) from SCZ, BPD and control subjects, and identified differentially expressed genes in these disorders. We found 105 and 153 genes differentially expressed in SCZ and BPD, respectively. By comparing the t-test scores, we found that many of the genes differentially expressed in SCZ and BPD are concordant in their expression level (q ⩽ 0.01, 53 genes; q ⩽ 0.05, 213 genes; q ⩽ 0.1, 885 genes). Using genome-wide association data from the Psychiatric Genomics Consortium, we found that these differentially and concordantly expressed genes were enriched in association signals for both SCZ (Po10 − 7) and BPD (P = 0.029). To our knowledge, this is the first time that a substantially large number of genes show concordant expression and association for both SCZ and BPD. Pathway analyses of these genes indicated that they are involved in the lysosome, Fc gamma receptor-mediated phagocytosis, regulation of actin cytoskeleton pathways, along with several cancer pathways. -

Role and Regulation of the P53-Homolog P73 in the Transformation of Normal Human Fibroblasts
Role and regulation of the p53-homolog p73 in the transformation of normal human fibroblasts Dissertation zur Erlangung des naturwissenschaftlichen Doktorgrades der Bayerischen Julius-Maximilians-Universität Würzburg vorgelegt von Lars Hofmann aus Aschaffenburg Würzburg 2007 Eingereicht am Mitglieder der Promotionskommission: Vorsitzender: Prof. Dr. Dr. Martin J. Müller Gutachter: Prof. Dr. Michael P. Schön Gutachter : Prof. Dr. Georg Krohne Tag des Promotionskolloquiums: Doktorurkunde ausgehändigt am Erklärung Hiermit erkläre ich, dass ich die vorliegende Arbeit selbständig angefertigt und keine anderen als die angegebenen Hilfsmittel und Quellen verwendet habe. Diese Arbeit wurde weder in gleicher noch in ähnlicher Form in einem anderen Prüfungsverfahren vorgelegt. Ich habe früher, außer den mit dem Zulassungsgesuch urkundlichen Graden, keine weiteren akademischen Grade erworben und zu erwerben gesucht. Würzburg, Lars Hofmann Content SUMMARY ................................................................................................................ IV ZUSAMMENFASSUNG ............................................................................................. V 1. INTRODUCTION ................................................................................................. 1 1.1. Molecular basics of cancer .......................................................................................... 1 1.2. Early research on tumorigenesis ................................................................................. 3 1.3. Developing -

Human Induced Pluripotent Stem Cell–Derived Podocytes Mature Into Vascularized Glomeruli Upon Experimental Transplantation
BASIC RESEARCH www.jasn.org Human Induced Pluripotent Stem Cell–Derived Podocytes Mature into Vascularized Glomeruli upon Experimental Transplantation † Sazia Sharmin,* Atsuhiro Taguchi,* Yusuke Kaku,* Yasuhiro Yoshimura,* Tomoko Ohmori,* ‡ † ‡ Tetsushi Sakuma, Masashi Mukoyama, Takashi Yamamoto, Hidetake Kurihara,§ and | Ryuichi Nishinakamura* *Department of Kidney Development, Institute of Molecular Embryology and Genetics, and †Department of Nephrology, Faculty of Life Sciences, Kumamoto University, Kumamoto, Japan; ‡Department of Mathematical and Life Sciences, Graduate School of Science, Hiroshima University, Hiroshima, Japan; §Division of Anatomy, Juntendo University School of Medicine, Tokyo, Japan; and |Japan Science and Technology Agency, CREST, Kumamoto, Japan ABSTRACT Glomerular podocytes express proteins, such as nephrin, that constitute the slit diaphragm, thereby contributing to the filtration process in the kidney. Glomerular development has been analyzed mainly in mice, whereas analysis of human kidney development has been minimal because of limited access to embryonic kidneys. We previously reported the induction of three-dimensional primordial glomeruli from human induced pluripotent stem (iPS) cells. Here, using transcription activator–like effector nuclease-mediated homologous recombination, we generated human iPS cell lines that express green fluorescent protein (GFP) in the NPHS1 locus, which encodes nephrin, and we show that GFP expression facilitated accurate visualization of nephrin-positive podocyte formation in -

Integrative Analysis of Disease Signatures Shows Inflammation Disrupts Juvenile Experience-Dependent Cortical Plasticity
New Research Development Integrative Analysis of Disease Signatures Shows Inflammation Disrupts Juvenile Experience- Dependent Cortical Plasticity Milo R. Smith1,2,3,4,5,6,7,8, Poromendro Burman1,3,4,5,8, Masato Sadahiro1,3,4,5,6,8, Brian A. Kidd,2,7 Joel T. Dudley,2,7 and Hirofumi Morishita1,3,4,5,8 DOI:http://dx.doi.org/10.1523/ENEURO.0240-16.2016 1Department of Neuroscience, Icahn School of Medicine at Mount Sinai, New York, New York 10029, 2Department of Genetics and Genomic Sciences, Icahn School of Medicine at Mount Sinai, New York, New York 10029, 3Department of Psychiatry, Icahn School of Medicine at Mount Sinai, New York, New York 10029, 4Department of Ophthalmology, Icahn School of Medicine at Mount Sinai, New York, New York 10029, 5Mindich Child Health and Development Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, 6Graduate School of Biomedical Sciences, Icahn School of Medicine at Mount Sinai, New York, New York 10029, 7Icahn Institute for Genomics and Multiscale Biology, Icahn School of Medicine at Mount Sinai, New York, New York 10029, and 8Friedman Brain Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029 Visual Abstract Throughout childhood and adolescence, periods of heightened neuroplasticity are critical for the development of healthy brain function and behavior. Given the high prevalence of neurodevelopmental disorders, such as autism, identifying disruptors of developmental plasticity represents an essential step for developing strategies for prevention and intervention. Applying a novel computational approach that systematically assessed connections between 436 transcriptional signatures of disease and multiple signatures of neuroplasticity, we identified inflammation as a common pathological process central to a diverse set of diseases predicted to dysregulate Significance Statement During childhood and adolescence, heightened neuroplasticity allows the brain to reorganize and adapt to its environment. -

CDC42EP1 Sirna (Rat)
For research purposes only, not for human use Product Data Sheet CDC42EP1 siRNA (Rat) Catalog # Source Reactivity Applications CRR5514 Synthetic R RNAi Description siRNA to inhibit CDC42EP1 expression using RNA interference Specificity CDC42EP1 siRNA (Rat) is a target-specific 19-23 nt siRNA oligo duplexes designed to knock down gene expression. Form Lyophilized powder Gene Symbol CDC42EP1 Alternative Names BORG5; Cdc42 effector protein 1; Binder of Rho GTPases 5 Entrez Gene 315121 (Rat) SwissProt A1A5P0 (Rat) Purity > 97% Quality Control Oligonucleotide synthesis is monitored base by base through trityl analysis to ensure appropriate coupling efficiency. The oligo is subsequently purified by affinity-solid phase extraction. The annealed RNA duplex is further analyzed by mass spectrometry to verify the exact composition of the duplex. Each lot is compared to the previous lot by mass spectrometry to ensure maximum lot-to-lot consistency. Components We offers pre-designed sets of 3 different target-specific siRNA oligo duplexes of rat CDC42EP1 gene. Each vial contains 5 nmol of lyophilized siRNA. The duplexes can be transfected individually or pooled together to achieve knockdown of the target gene, which is most commonly assessed by qPCR or western blot. Our siRNA oligos are also chemically modified (2’-OMe) at no extra charge for increased stability and enhanced knockdown in vitro and in vivo. Directions for Use We recommends transfection with 100 nM siRNA 48 to 72 hours prior to cell lysis. Application key: E- ELISA, WB- Western blot, -
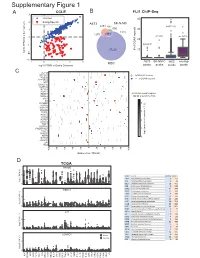
Supplementary Figure 1
6XSSOHPHQWDU\)LJXUH CCLE B FLI1 ChIP-Seq A 3 FRPPRQ 40 (ZLQJ6SHFLILF 2 A673 6.10& 477 Q 690 30 0 UHSHDWV Q Q A 20 í Q 57226 RI**$ ORJ)3.0LQRWKHUFDQFHUV í í A673 6.10& 06& RYHUODS í 0 23 06& ORJ)3.0LQ(ZLQJ6DUFRPD SHDNV SHDNV SHDNV SHDNV & ./) 67($3 (:6)/,ERXQG 3'(% ! **$AUHSHDWV 3335$ ABI3 '1$-& '863 13<5 .&1( 5%0 DOO**$AUHSHDWUHJLRQV 1.; NEDURXQGWKH766 9$9 ;* .&1$% 4 .&1$ 51) 0<20 3 355/ 355T4 &'$ $'5% 2 '&'& 711, 8*7$ 3+263+2 )(=) *1*7 ORJ QXPEHURIUHSHDWV A571 0 13<5 /2;+' /,3, 5$; 0 40 60 20 GLVWDQFHIURP766 NE D TCGA 35$0( 5 code cancer normal tumor LAML Acute Myeloid Leukemia 0 151 ORJ )3.0 ACC AdrenocoƌƟcal carcinoma 0 79 0 BLCA Bladder Urothelial Carcinoma 19 411 LGG Brain Lower Grade Glioma 0 529 COAD Colon adenocarcinoma 41 464 5%0 ESCA Esophageal carcinoma 11 162 GBM Glioblastoma muůƟforme 5 168 5 KICH Kidney Chromophobe 24 65 KIRP Kidney renal papillary cell carcinoma 32 289 LIHC Liver hepatocellular carcinoma 50 374 ORJ )3.0 LUAD Lung adenocarcinoma 59 526 0 LUSC Lung squamous cell carcinoma 49 501 DLBC Lymphoid Neoplasm Diīuse Large B-cell Lym 042 /,3, MESO Mesothelioma 0 1 OV Ovarian serous cystadenocarcinoma 0 379 PAAD PancreĂƟc adenocarcinoma 4 178 5 PCPG Pheochromocytoma and Paraganglioma 3 150 PRAD Prostate adenocarcinoma 52 499 READ Rectum adenocarcinoma 1 6 ORJ )3.0 SARC Sarcoma 0 35 0 SKCM Skin Cutaneous Melanoma 1 471 STAD Stomach adenocarcinoma 32 375 /2;+' WXPRU TGCT dĞƐƟĐƵůĂƌ'ĞƌŵĞůůdƵŵŽƌƐ 0156 THCA Thyroid carcinoma 58 510 QRUPDO UCS Uterine Carcinosarcoma 0 56 5 ORJ )3.0 0 / / 29 $&& 8&6 /** *%0 -

CREB-Dependent Transcription in Astrocytes: Signalling Pathways, Gene Profiles and Neuroprotective Role in Brain Injury
CREB-dependent transcription in astrocytes: signalling pathways, gene profiles and neuroprotective role in brain injury. Tesis doctoral Luis Pardo Fernández Bellaterra, Septiembre 2015 Instituto de Neurociencias Departamento de Bioquímica i Biologia Molecular Unidad de Bioquímica y Biologia Molecular Facultad de Medicina CREB-dependent transcription in astrocytes: signalling pathways, gene profiles and neuroprotective role in brain injury. Memoria del trabajo experimental para optar al grado de doctor, correspondiente al Programa de Doctorado en Neurociencias del Instituto de Neurociencias de la Universidad Autónoma de Barcelona, llevado a cabo por Luis Pardo Fernández bajo la dirección de la Dra. Elena Galea Rodríguez de Velasco y la Dra. Roser Masgrau Juanola, en el Instituto de Neurociencias de la Universidad Autónoma de Barcelona. Doctorando Directoras de tesis Luis Pardo Fernández Dra. Elena Galea Dra. Roser Masgrau In memoriam María Dolores Álvarez Durán Abuela, eres la culpable de que haya decidido recorrer el camino de la ciencia. Que estas líneas ayuden a conservar tu recuerdo. A mis padres y hermanos, A Meri INDEX I Summary 1 II Introduction 3 1 Astrocytes: physiology and pathology 5 1.1 Anatomical organization 6 1.2 Origins and heterogeneity 6 1.3 Astrocyte functions 8 1.3.1 Developmental functions 8 1.3.2 Neurovascular functions 9 1.3.3 Metabolic support 11 1.3.4 Homeostatic functions 13 1.3.5 Antioxidant functions 15 1.3.6 Signalling functions 15 1.4 Astrocytes in brain pathology 20 1.5 Reactive astrogliosis 22 2 The transcription -

The Chromatin Remodelling Component SMARCB1/INI1
Pancione et al. Journal of Translational Medicine 2013, 11:297 http://www.translational-medicine.com/content/11/1/297 RESEARCH Open Access The chromatin remodelling component SMARCB1/INI1 influences the metastatic behavior of colorectal cancer through a gene signature mapping to chromosome 22 Massimo Pancione1*†, Andrea Remo2†, Caterina Zanella2, Lina Sabatino1, Arturo Di Blasi3, Carmelo Laudanna1, Laura Astati2, Michele Rocco4, Delfina Bifano4, Paolo Piacentini2, Laura Pavan2, Alberto Purgato2, Filippo Greco2, Alberto Talamini2, Andrea Bonetti2, Michele Ceccarelli1, Roberto Vendraminelli2, Erminia Manfrin5 and Vittorio Colantuoni1* Background: INI1 (Integrase interactor 1), also known as SMARCB1, is the most studied subunit of chromatin remodelling complexes. Its role in colorectal tumorigenesis is not known. Methods: We examined SMARCB1/INI1 protein expression in 134 cases of colorectal cancer (CRC) and 60 matched normal mucosa by using tissue microarrays and western blot and categorized the results according to mismatch repair status (MMR), CpG island methylator phenotype, biomarkers of tumor differentiation CDX2, CK20, vimentin and p53. We validated results in two independent data sets and in cultured CRC cell lines. Results: Herein, we show that negative SMARCB1/INI1 expression (11% of CRCs) associates with loss of CDX2, poor differentiation, liver metastasis and shorter patients’ survival regardless of the MMR status or tumor stage. Unexpectedly, even CRCs displaying diffuse nuclear INI1 staining (33%) show an adverse prognosis and vimentin over-expression, in comparison with the low expressing group (56%). The negative association of SMARCB1/INI1-lack of expression with a metastatic behavior is enhanced by the TP53 status. By interrogating global gene expression from two independent cohorts of 226 and 146 patients, weconfirmtheprognosticresults and identify a gene signature characterized by SMARCB1/INI1 deregulation. -

Agricultural University of Athens
ΓΕΩΠΟΝΙΚΟ ΠΑΝΕΠΙΣΤΗΜΙΟ ΑΘΗΝΩΝ ΣΧΟΛΗ ΕΠΙΣΤΗΜΩΝ ΤΩΝ ΖΩΩΝ ΤΜΗΜΑ ΕΠΙΣΤΗΜΗΣ ΖΩΙΚΗΣ ΠΑΡΑΓΩΓΗΣ ΕΡΓΑΣΤΗΡΙΟ ΓΕΝΙΚΗΣ ΚΑΙ ΕΙΔΙΚΗΣ ΖΩΟΤΕΧΝΙΑΣ ΔΙΔΑΚΤΟΡΙΚΗ ΔΙΑΤΡΙΒΗ Εντοπισμός γονιδιωματικών περιοχών και δικτύων γονιδίων που επηρεάζουν παραγωγικές και αναπαραγωγικές ιδιότητες σε πληθυσμούς κρεοπαραγωγικών ορνιθίων ΕΙΡΗΝΗ Κ. ΤΑΡΣΑΝΗ ΕΠΙΒΛΕΠΩΝ ΚΑΘΗΓΗΤΗΣ: ΑΝΤΩΝΙΟΣ ΚΟΜΙΝΑΚΗΣ ΑΘΗΝΑ 2020 ΔΙΔΑΚΤΟΡΙΚΗ ΔΙΑΤΡΙΒΗ Εντοπισμός γονιδιωματικών περιοχών και δικτύων γονιδίων που επηρεάζουν παραγωγικές και αναπαραγωγικές ιδιότητες σε πληθυσμούς κρεοπαραγωγικών ορνιθίων Genome-wide association analysis and gene network analysis for (re)production traits in commercial broilers ΕΙΡΗΝΗ Κ. ΤΑΡΣΑΝΗ ΕΠΙΒΛΕΠΩΝ ΚΑΘΗΓΗΤΗΣ: ΑΝΤΩΝΙΟΣ ΚΟΜΙΝΑΚΗΣ Τριμελής Επιτροπή: Aντώνιος Κομινάκης (Αν. Καθ. ΓΠΑ) Ανδρέας Κράνης (Eρευν. B, Παν. Εδιμβούργου) Αριάδνη Χάγερ (Επ. Καθ. ΓΠΑ) Επταμελής εξεταστική επιτροπή: Aντώνιος Κομινάκης (Αν. Καθ. ΓΠΑ) Ανδρέας Κράνης (Eρευν. B, Παν. Εδιμβούργου) Αριάδνη Χάγερ (Επ. Καθ. ΓΠΑ) Πηνελόπη Μπεμπέλη (Καθ. ΓΠΑ) Δημήτριος Βλαχάκης (Επ. Καθ. ΓΠΑ) Ευάγγελος Ζωίδης (Επ.Καθ. ΓΠΑ) Γεώργιος Θεοδώρου (Επ.Καθ. ΓΠΑ) 2 Εντοπισμός γονιδιωματικών περιοχών και δικτύων γονιδίων που επηρεάζουν παραγωγικές και αναπαραγωγικές ιδιότητες σε πληθυσμούς κρεοπαραγωγικών ορνιθίων Περίληψη Σκοπός της παρούσας διδακτορικής διατριβής ήταν ο εντοπισμός γενετικών δεικτών και υποψηφίων γονιδίων που εμπλέκονται στο γενετικό έλεγχο δύο τυπικών πολυγονιδιακών ιδιοτήτων σε κρεοπαραγωγικά ορνίθια. Μία ιδιότητα σχετίζεται με την ανάπτυξη (σωματικό βάρος στις 35 ημέρες, ΣΒ) και η άλλη με την αναπαραγωγική -

The Borg Family of Cdc42 Effector Proteins Cdc42ep1–5
Biochemical Society Transactions (2016) 44 1709–1716 DOI: 10.1042/BST20160219 The Borg family of Cdc42 effector proteins Cdc42EP1–5 Aaron J. Farrugia and Fernando Calvo Tumour Microenvironment Team, Division of Cancer Biology, Institute of Cancer Research, 237 Fulham Road, London SW2 6JB, U.K. Correspondence: Fernando Calvo ([email protected]) Despite being discovered more than 15 years ago, the Borg (binder of Rho GTPases) family of Cdc42 effector proteins (Cdc42EP1–5) remains largely uncharacterised and rela- tively little is known about their structure, regulation and role in development and disease. Recent studies are starting to unravel some of the key functional and mechanistic aspects of the Borg proteins, including their role in cytoskeletal remodelling and signal- ling. In addition, the participation of Borg proteins in important cellular processes such as cell shape, directed migration and differentiation is slowly emerging, directly linking Borgs with important physiological and pathological processes such as angiogenesis, neuro- transmission and cancer-associated desmoplasia. Here, we review some of these find- ings and discuss future prospects. Introduction The Rho GTPase family member Cdc42 regulates a diverse range of cellular functions including cyto- kinesis, cytoskeletal remodelling and cell polarity [1,2]. Like other Rho family members, Cdc42 cycles between two tightly regulated conformational states, a GTP-bound active state and a GDP-bound inactive state [3]. Activated Cdc42 exerts its functions by interacting with downstream effectors con- taining a Cdc42/Rac interactive binding (CRIB) motif [3,4]. Several Cdc42 effector proteins, including kinases and scaffolds, have been well characterised [5]; however, the functions of others remain rela- tively unknown.