Dissecting the Role of Ubiquitylation in the DNA Damage Response Checkpoint in G2
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Bioinformatics-Based Screening of Key Genes for Transformation of Liver
Jiang et al. J Transl Med (2020) 18:40 https://doi.org/10.1186/s12967-020-02229-8 Journal of Translational Medicine RESEARCH Open Access Bioinformatics-based screening of key genes for transformation of liver cirrhosis to hepatocellular carcinoma Chen Hao Jiang1,2, Xin Yuan1,2, Jiang Fen Li1,2, Yu Fang Xie1,2, An Zhi Zhang1,2, Xue Li Wang1,2, Lan Yang1,2, Chun Xia Liu1,2, Wei Hua Liang1,2, Li Juan Pang1,2, Hong Zou1,2, Xiao Bin Cui1,2, Xi Hua Shen1,2, Yan Qi1,2, Jin Fang Jiang1,2, Wen Yi Gu4, Feng Li1,2,3 and Jian Ming Hu1,2* Abstract Background: Hepatocellular carcinoma (HCC) is the most common type of liver tumour, and is closely related to liver cirrhosis. Previous studies have focussed on the pathogenesis of liver cirrhosis developing into HCC, but the molecular mechanism remains unclear. The aims of the present study were to identify key genes related to the transformation of cirrhosis into HCC, and explore the associated molecular mechanisms. Methods: GSE89377, GSE17548, GSE63898 and GSE54236 mRNA microarray datasets from Gene Expression Omni- bus (GEO) were analysed to obtain diferentially expressed genes (DEGs) between HCC and liver cirrhosis tissues, and network analysis of protein–protein interactions (PPIs) was carried out. String and Cytoscape were used to analyse modules and identify hub genes, Kaplan–Meier Plotter and Oncomine databases were used to explore relationships between hub genes and disease occurrence, development and prognosis of HCC, and the molecular mechanism of the main hub gene was probed using Kyoto Encyclopedia of Genes and Genomes(KEGG) pathway analysis. -

The Regulatory Roles of Phosphatases in Cancer
Oncogene (2014) 33, 939–953 & 2014 Macmillan Publishers Limited All rights reserved 0950-9232/14 www.nature.com/onc REVIEW The regulatory roles of phosphatases in cancer J Stebbing1, LC Lit1, H Zhang, RS Darrington, O Melaiu, B Rudraraju and G Giamas The relevance of potentially reversible post-translational modifications required for controlling cellular processes in cancer is one of the most thriving arenas of cellular and molecular biology. Any alteration in the balanced equilibrium between kinases and phosphatases may result in development and progression of various diseases, including different types of cancer, though phosphatases are relatively under-studied. Loss of phosphatases such as PTEN (phosphatase and tensin homologue deleted on chromosome 10), a known tumour suppressor, across tumour types lends credence to the development of phosphatidylinositol 3--kinase inhibitors alongside the use of phosphatase expression as a biomarker, though phase 3 trial data are lacking. In this review, we give an updated report on phosphatase dysregulation linked to organ-specific malignancies. Oncogene (2014) 33, 939–953; doi:10.1038/onc.2013.80; published online 18 March 2013 Keywords: cancer; phosphatases; solid tumours GASTROINTESTINAL MALIGNANCIES abs in sera were significantly associated with poor survival in Oesophageal cancer advanced ESCC, suggesting that they may have a clinical utility in Loss of PTEN (phosphatase and tensin homologue deleted on ESCC screening and diagnosis.5 chromosome 10) expression in oesophageal cancer is frequent, Cao et al.6 investigated the role of protein tyrosine phosphatase, among other gene alterations characterizing this disease. Zhou non-receptor type 12 (PTPN12) in ESCC and showed that PTPN12 et al.1 found that overexpression of PTEN suppresses growth and protein expression is higher in normal para-cancerous tissues than induces apoptosis in oesophageal cancer cell lines, through in 20 ESCC tissues. -

Lenalidomide Induces Cell Death in an MDS-Derived Cell Line with Deletion of Chromosome 5Q by Inhibition of Cytokinesis
Leukemia (2010) 24, 748–755 & 2010 Macmillan Publishers Limited All rights reserved 0887-6924/10 $32.00 www.nature.com/leu ORIGINAL ARTICLE Lenalidomide induces cell death in an MDS-derived cell line with deletion of chromosome 5q by inhibition of cytokinesis A Matsuoka1, A Tochigi1, M Kishimoto1, T Nakahara1, T Kondo1, T Tsujioka1, T Tasaka1, Y Tohyama2 and K Tohyama1 1Department of Laboratory Medicine, Kawasaki Medical School, Okayama, Japan; and 2Division of Biochemistry, Faculty of Pharmaceutical Sciences, Himeji Dokkyo University, Hyogo, Japan Myelodysplastic syndromes (MDS) are a group of hematopoietic higher-risk groups with del(5q) that are susceptible to leukemic stem cell disorders characterized by refractory cytopenias transformation.6 Several hematopoiesis-related genes and tumor and susceptibility to leukemic transformation. On a subset of suppressor genes are located at 5q locus, and SPARC,7 MDS patients with deletion of the long arm of chromosome5 8 9 10 11 (del(5q)), lenalidomide exerts hematological and cytogenetic CTNNA1, EGR1, RPS14 and CDC25C are reported as effects, but the underlying pharmacological mechanisms are the candidate genes of MDS with del(5q) or 5q- syndrome. not fully understood. In this study, we have investigated the Lenalidomide, a derivative of thalidomide, is shown to in vitro effects of lenalidomide on an MDS-derived cell line, exert plenty of biological actions including inhibition of MDS-L, which carries del(5q) and complex chromosome angiogenesis,12 suppression of proinflammatory cytokine abnormalities. We found that the growth of MDS-L cells was production such as TNF-a,13 enhancement of T- and NK-cell specifically suppressed mainly by apoptosis, and in addition, 14–16 multinucleated cells were frequently formed and finally died activation. -

Identification of Seven Hub Genes As the Novel Biomarkers in Triple
Identication of Seven Hub Genes as the Novel Biomarkers in Triple-negative Breast Cancer and Breast Cancer Metastasis Huanxian Wu Southern Medical University Huining Lian Southern Medical University Nanfang Hospital Qianqing Chen Southern Medical University Jinlamao Yang Southern Medical University Nanfang Hospital Baofang Ou Southern Medical University Dongling Quan Southern Medical University Lei Zhou Southern Medical University Lin Lv Southern Medical University Minfeng Liu Southern Medical University Nanfang Hospital Shaoyu Wu ( [email protected] ) Guangdong Provincial Key Laboratory of New Drug Screening, School of Pharmaceutical Science, Southern Medical University, Guangzhou, Guangdong, 510515, PR China. https://orcid.org/0000-0002- 1247-5295 Research article Keywords: Triple-negative breast cancer, Metastasis, Biomarkers, Prognostic signature Posted Date: October 5th, 2020 DOI: https://doi.org/10.21203/rs.3.rs-73076/v1 Page 1/20 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 2/20 Abstract Background: Breast cancer is one of the most common malignant tumors with the highest morbidity and mortality among women. Compared with the other breast cancer subtypes, Triple-negative breast cancer (TNBC) has a higher probability of recurrence and is prone to distant metastasis. To reveal the underlying disease mechanisms and identify more effective biomarkers for TNBC and breast cancer metastasis. Methods: Gene Ontology and KEGG pathway analysis were used for investigating the role of overlapping differentially expressed genes (DEGs). Hub genes among these DEGs were determined by the protein- protein interactions network analysis and CytoHubba. Oncomine databases were used for verifying the clinical relevance of hub genes. -

CDKN3 Mrna As a Biomarker for Survival and Therapeutic Target in Cervical Cancer
RESEARCH ARTICLE CDKN3 mRNA as a Biomarker for Survival and Therapeutic Target in Cervical Cancer Eira Valeria Barrón1,2, Edgar Roman-Bassaure3, Ana Laura Sánchez-Sandoval4, Ana María Espinosa1, Mariano Guardado-Estrada1, Ingrid Medina1, Eligia Juárez1, Ana Alfaro1, Miriam Bermúdez1, Rubén Zamora5,6, Carlos García-Ruiz1, Juan Carlos Gomora4, Susana Kofman7, E. Martha Pérez-Armendariz2¤, Jaime Berumen1,2* 1 Unidad de Medicina Genómica, Facultad de Medicina, Universidad Nacional Autónoma de México/ Hospital General de México, México City, México, 2 Departamento de Medicina Experimental, Facultad de Medicina, Universidad Nacional Autónoma de México, México City, México, 3 Servicio de Oncología, Hospital General de México, México City, México, 4 Departamento de Neuropatología Molecular, División de Neurociencias, Instituto de Fisiología Celular, Universidad Nacional Autónoma de México, México City, México, 5 Departamento de Inmunología, Instituto de Investigaciones Biomédicas, Universidad Nacional Autónoma de México, México City, México, 6 Laboratorio de Biología Molecular, Asociación para Evitar la Ceguera en México Hospital Dr. Luis Sánchez-Bulnes, México City, México, 7 Servicio de Genética, Hospital General de México/Facultad de Medicina, Universidad Nacional Autónoma de México, México City, México ¤ Current address: Departamento de Biología, Celular y Tisular, Facultad de Medicina, Universidad Nacional OPEN ACCESS Autónoma de México, México City, México * [email protected] Citation: Barrón EV, Roman-Bassaure E, Sánchez- Sandoval AL, Espinosa AM, Guardado-Estrada M, Medina I, et al. (2015) CDKN3 mRNA as a Biomarker for Survival and Therapeutic Target in Cervical Abstract Cancer. PLoS ONE 10(9): e0137397. doi:10.1371/ CDKN3 journal.pone.0137397 The cyclin-dependent kinase inhibitor 3 ( ) gene, involved in mitosis, is upregulated in cervical cancer (CC). -

PTEN Enhances G2/M Arrest in Etoposide-Treated MCF-7 Cells
ONCOLOGY REPORTS 35: 2707-2714, 2016 PTEN enhances G2/M arrest in etoposide-treated MCF-7 cells through activation of the ATM pathway RUOPENG ZHANG1,2, LI ZHU2, LIRONG ZHANG2, ANLI XU2, ZHENGWEI LI3, YIJUAN XU3, PEI HE3, MAOQING WU4, FENGXIANG WEI5 and CHENHONG Wang1 1Department of Obstetrics and Gynecology, Shenzhen Maternity and Child Healthcare Hospital, Affiliated to Southern Medical University, Longgang, Shenzhen, Guangdong 518028; 2Department of Reproductive Medicine, Affiliated Hospital of Dali University, Dali, Yunnan 671000;3 Clinical Medicine College of Dali University, Dali, Yunnan 671000, P.R. China; 4Renal Division, Department of Medicine, Brigham and Women's Hospital, Harvard Medical School, Boston, MA 02115, USA; 5The Genetics Laboratory, Shenzhen Longgang District Maternity and Child Healthcare Hospital, Longgang, Shenzhen, Guangdong 518028, P.R. China Received November 19, 2015; Accepted December 27, 2015 DOI: 10.3892/or.2016.4674 Abstract. As an effective tumor suppressor, phosphatase and role in etoposide-induced G2/M arrest by facilitating the acti- tensin homolog (PTEN) has attracted the increased attention of vation of the ATM pathway, and PTEN was required for the scientists. Recent studies have shown that PTEN plays unique proper activation of checkpoints in response to DNA damage roles in the DNA damage response (DDR) and can interact in MCF-7 cells. with the Chk1 pathway. However, little is known about how PTEN contributes to DDR through the ATM-Chk2 pathway. It Introduction is well-known that etoposide induces G2/M arrest in a variety of cell lines, including MCF-7 cells. The DNA damage-induced Cellular DNA is constantly challenged by either endogenous G2/M arrest results from the activation of protein kinase [reactive oxygen species (ROS)] or exogenous (UV) factors. -

A Novel Synthetic Inhibitor of CDC25 Phosphatases: BN82002
[CANCER RESEARCH 64, 3320–3325, May 1, 2004] A Novel Synthetic Inhibitor of CDC25 Phosphatases: BN82002 Marie-Christine Brezak,1 Muriel Quaranta,2 Odile Monde´sert,2 Marie-Odile Galcera,1 Olivier Lavergne,1 Fre´de´ric Alby,2 Martine Cazales,2 Ve´ronique Baldin,2 Christophe Thurieau,1 Jeremiath Harnett,1 Christophe Lanco,1 Philip G. Kasprzyk,1,3 Gregoire P. Prevost,1 and Bernard Ducommun2 1IPSEN, Institut Henri Beaufour, Les Ulis Cedex, France; 2Laboratoire de Biologie Cellulaire et Moleculaire du Controle de la Proliferation-Centre National de la Recherche Scientifique UMR5088-IFR109 “Institut d’Exploration Fonctionnelle des Ge´nomes,” Universite´ Paul Sabatier, Toulouse, France; and 3IPSEN, Biomeasure, Milford, Massachusetts ABSTRACT B at mitosis (2) where it dephosphorylates tyrosine 15 and threonine 14. It also plays a role in the control of the initiation of S phase (5). CDC25 dual-specificity phosphatases are essential regulators that de- The elucidation of the specific role of each isoform at specific stages phosphorylate and activate cyclin-dependent kinase/cyclin complexes at of the cell cycle is a major issue that is still currently under investi- key transitions of the cell cycle. CDC25 activity is currently considered to be an interesting target for the development of new antiproliferative gation. agents. Here we report the identification of a new CDC25 inhibitor and Overexpression of CDC25A and B, but not C, has been observed in the characterization of its effects at the molecular and cellular levels, and a variety of cancers (i.e., breast, ovary, head and neck, and colon) with in animal models. a striking association with tumor aggressiveness and poor prognosis BN82002 inhibits the phosphatase activity of recombinant human (6–9). -
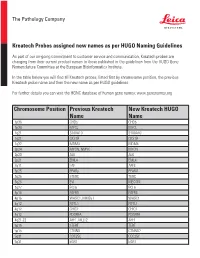
Chromosome Position Previous Kreatech Name New Kreatech
The Pathology Company Kreatech Probes assigned new names as per HUGO Naming Guidelines As part of our on-going commitment to customer service and communication, Kreatech probes are changing from their current product names to those published in the guidelines from the HUGO Gene Nomenclature Committee at the European Bioinformatics Institute. In the table below you will find all Kreatech probes, listed first by chromosome position, the previous Kreatech probe name and then the new name as per HUGO guidelines. For further details you can visit the HGNC database of human gene names: www.genenames.org Chromosome Position Previous Kreatech New Kreatech HUGO Name Name 1p36 CHD5 CHD5 1p34 MYCL MYCL 1q21 S100A10 S100A10 1q21 CKS1B CKS1B 1q32 MDM4 MDM4 2p24 MYCN, NMYC MYCN 2p23 ALK ALK 2p21 EML4 EML4 2q11 LAF AFF3 3p25 PPARy PPARG 3q26 hTERC TERC 3q26 EVI MECOM 3q27 BCL6 BCL6 4p16 FGFR3 FGFR3 4p16 WHSC1, MMSET WHSC1 4q12 FIP1L1 FIP1L1 4q12 CHIC2 CHIC2 4q12 PDGFRA PDGFRA 4q21-22 AFF1, MLLT2 AFF1 5p15 hTERT TERT 5p15 CTNND CTNND2 5q31 CDC25C CDC25C 5q31 EGR1 EGR1 The Pathology Company 5q33 PDGFRB PDGFRB 5q33 CSF1R CSF1R 5q33 RPS14 RPS14 5q35 NSD1 NSD1 5q35 FGFR4 FGFR4 6p25 DUSP22 DUSP22 6p25 IRF4 IRF4 6p24 RREB1 RREB1 6p23 MYB MYB 6p22 DEK DEK 6p21 CCND3 CCND3 6q21 SEC63 SEC63 6q22 ROS1 ROS1 6q27 MLLT4, AF6 MLLT4 7p11 EGFR, Her1 EGFR 7q11 ELN ELN 7q11 LIMK1 LIMK1 7q22 CUTL1 CUX1 7q31 C-MET MET 7q31 MDFIC MDFIC 8p23 GATA4 GATA4 8p21 PNOC PNOC 8p11 FGFR1 FGFR1 8q21 ETO RUNX1T 8q24 C-MYC MYC 9p24 JAK2 JAK2 9p21 MLLT3, AF9 MLLT3 9p21 p16 CDKN2A 9q34 -

Direct Roles of the Signaling Kinase RSK2 in Cdc25c Activation During Xenopus Oocyte Maturation
Direct roles of the signaling kinase RSK2 in Cdc25C activation during Xenopus oocyte maturation Ruoning Wanga,b,1, Sung Yun Jungc, Chuan Fen Wua, Jun Qinc, Ryuji Kobayashid, Gary E. Gallicke, and Jian Kuanga,b,2 Departments of aExperimental Therapeutics, dMolecular Pathology, and eGenitourinary Medical Oncology, University of Texas M. D. Anderson Cancer Center, Houston, TX 77030; bProgram in Genes and Development, University of Texas Graduate School of Biomedical Sciences, Houston, TX 77030; and cDepartment of Biochemistry, Baylor College of Medicine, Houston, TX 77030 Edited* by John Gerhart, University of California, Berkeley, CA, and approved October 6, 2010 (received for review March 22, 2010) The induction of M phase in eukaryotic cell cycles requires robust phosphorylations catalyzed by such identified kinases “primary activation of Cdc2/cyclin B by Cdc25, which itself is robustly activated phosphorylations.” By fractionation of MEE, our previous results by serine/threonine phosphorylations. Although multiple protein demonstrated that 10–20% of the primary Cdc25C phosphory- kinases that directly activate Cdc25C have been identified, whether lating activity is due to Cdc2/cyclin B, which phosphorylates the combination of different primary phosphorylations of Cdc25C Cdc25C at the proline-directed sites T138, S285, and T308 and is sufficient to fully activate Cdc25C has not been determined. By activates GST-Cdc25C two- to fourfold. Approximately 40% of analyzing the GST-Cdc25C phosphorylating activity in Xenopus egg the activity is due to p42 MAPK, which phosphorylates Cdc25C at extracts, we previously defined roles of MAPK and Cdc2/cyclin B in the proline-directed sites T48, T138, and S205 and also activates partially activating Cdc25C and predicted the presence of another GST-Cdc25C two- to fourfold. -

CDC25 Phosphatases in Cancer Cells…
REVIEWS CDC25 phosphatases in cancer cells: key players? Good targets? Rose Boutros*, Valérie Lobjois* and Bernard Ducommun*‡ Abstract | Cell division cycle 25 (CDC25) phosphatases regulate key transitions between cell cycle phases during normal cell division, and in the event of DNA damage they are key targets of the checkpoint machinery that ensures genetic stability. Taking only this into consideration, it is not surprising that CDC25 overexpression has been reported in a significant number of human cancers. However, in light of the significant body of evidence detailing the stringent complexity with which CDC25 activities are regulated, the significance of CDC25 overexpression in a subset of cancers and its association with poor prognosis are proving difficult to assess. We will focus on the roles of CDC25 phosphatases in both normal and abnormal cell proliferation, provide a critical assessment of the current data on CDC25 overexpression in cancer, and discuss both current and future therapeutic strategies for targeting CDC25 activity in cancer treatment. Dual-specificity protein The cell division cycle 25 (CDC25) family of proteins are Among the different species, the catalytic domains of phosphatase highly conserved dual specificity phosphatases that acti- CDC25 proteins are quite conserved compared with A phosphoprotein vate cyclin-dependent kinase (CDK) complexes, which in the regulatory regions, which are far more diverse and phosphatase that is able to turn regulate progression through the cell division cycle. further subjected to alternative splicing events that gen- hydrolyse the phosphate ester 8 bond on both a tyrosine and a CDC25 phosphatases are also key components of the erate at least two variants for CDC25A and five each for 9,10 8,11 threonine or serine residue on checkpoint pathways that become activated in the event of CDC25B and CDC25C (FIG. -

Live-Cell Imaging Rnai Screen Identifies PP2A–B55α and Importin-Β1 As Key Mitotic Exit Regulators in Human Cells
LETTERS Live-cell imaging RNAi screen identifies PP2A–B55α and importin-β1 as key mitotic exit regulators in human cells Michael H. A. Schmitz1,2,3, Michael Held1,2, Veerle Janssens4, James R. A. Hutchins5, Otto Hudecz6, Elitsa Ivanova4, Jozef Goris4, Laura Trinkle-Mulcahy7, Angus I. Lamond8, Ina Poser9, Anthony A. Hyman9, Karl Mechtler5,6, Jan-Michael Peters5 and Daniel W. Gerlich1,2,10 When vertebrate cells exit mitosis various cellular structures can contribute to Cdk1 substrate dephosphorylation during vertebrate are re-organized to build functional interphase cells1. This mitotic exit, whereas Ca2+-triggered mitotic exit in cytostatic-factor- depends on Cdk1 (cyclin dependent kinase 1) inactivation arrested egg extracts depends on calcineurin12,13. Early genetic studies in and subsequent dephosphorylation of its substrates2–4. Drosophila melanogaster 14,15 and Aspergillus nidulans16 reported defects Members of the protein phosphatase 1 and 2A (PP1 and in late mitosis of PP1 and PP2A mutants. However, the assays used in PP2A) families can dephosphorylate Cdk1 substrates in these studies were not specific for mitotic exit because they scored pro- biochemical extracts during mitotic exit5,6, but how this relates metaphase arrest or anaphase chromosome bridges, which can result to postmitotic reassembly of interphase structures in intact from defects in early mitosis. cells is not known. Here, we use a live-cell imaging assay and Intracellular targeting of Ser/Thr phosphatase complexes to specific RNAi knockdown to screen a genome-wide library of protein substrates is mediated by a diverse range of regulatory and targeting phosphatases for mitotic exit functions in human cells. We subunits that associate with a small group of catalytic subunits3,4,17. -

PTEN Insufficiency Modulates ER+ Breast Cancer Cell Cycle Progression and Increases Cell Growth in Vitro and in Vivo
Journal name: Drug Design, Development and Therapy Article Designation: Original Research Year: 2015 Volume: 9 Drug Design, Development and Therapy Dovepress Running head verso: Chiang et al Running head recto: PTEN insufficiency increases breast cancer growth open access to scientific and medical research DOI: http://dx.doi.org/10.2147/DDDT.S86184 Open Access Full Text Article ORIGINAL RESEARCH PTEN insufficiency modulates ER+ breast cancer cell cycle progression and increases cell growth in vitro and in vivo Kun-Chun Chiang1,4 Abstract: Phosphatase and tensin homolog (PTEN), a well-known tumor suppressor gene Huang-Yang Chen1 and frequently mutated or lost in breast cancer, possesses the negative regulation function over Shu-Yuan Hsu2 the PI3K/Akt/mTOR pathway. PTEN insufficiency has been associated with advanced breast Jong-Hwei S Pang3 cancer and poor prognosis of breast cancer patients. Recently, target therapies aimed at PI3K/ Shang-Yu Wang4 Akt/mTOR pathway to treat breast cancer have got popularity. However, the exact effect of Jun-Te Hsu4 PTEN on breast cancer cells is still not well understood. This study demonstrated that PTEN knockdown in MCF-7 cells strengthened the downstream gene expressions, including p-Akt, Ta-Sen Yeh4 p-ERK1/2, p-mTOR, p-p70s6k, and p-GSK3β. PTEN knockdown MCF-7 cells had increased Li-Wei Chen5 cell growth and Ki-67 expression. Further Western blot demonstrated that p27 was repressed 6 For personal use only. Sheng-Fong Kuo obviously with p21 slightly inhibited and CDK1, 2, 4, 6, cyclin A, and Cdc25C were upregulated 7 Chi-Chin Sun in MCF-7 PTEN knockdown cells, leading to the higher growth rate.