Acid and Polarity Effects in Benzene Photoreactions with Alkenes and Alkynes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

ECO-Ssls for Pahs
Ecological Soil Screening Levels for Polycyclic Aromatic Hydrocarbons (PAHs) Interim Final OSWER Directive 9285.7-78 U.S. Environmental Protection Agency Office of Solid Waste and Emergency Response 1200 Pennsylvania Avenue, N.W. Washington, DC 20460 June 2007 This page intentionally left blank TABLE OF CONTENTS 1.0 INTRODUCTION .......................................................1 2.0 SUMMARY OF ECO-SSLs FOR PAHs......................................1 3.0 ECO-SSL FOR TERRESTRIAL PLANTS....................................4 5.0 ECO-SSL FOR AVIAN WILDLIFE.........................................8 6.0 ECO-SSL FOR MAMMALIAN WILDLIFE..................................8 6.1 Mammalian TRV ...................................................8 6.2 Estimation of Dose and Calculation of the Eco-SSL ........................9 7.0 REFERENCES .........................................................16 7.1 General PAH References ............................................16 7.2 References Used for Derivation of Plant and Soil Invertebrate Eco-SSLs ......17 7.3 References Rejected for Use in Derivation of Plant and Soil Invertebrate Eco-SSLs ...............................................................18 7.4 References Used in Derivation of Wildlife TRVs .........................25 7.5 References Rejected for Use in Derivation of Wildlife TRV ................28 i LIST OF TABLES Table 2.1 PAH Eco-SSLs (mg/kg dry weight in soil) ..............................4 Table 3.1 Plant Toxicity Data - PAHs ..........................................5 Table 4.1 -

Acrylonitrile
Common Name: ACRYLONITRILE CAS Number: 107-13-1 DOT Number: UN 1093 (Inhibited) RTK Substance number: 0024 DOT Hazard Class: 3 (Flammable Liquid) Date: May 1998 Revisions: December 2005 ------------------------------------------------------------------------- ------------------------------------------------------------------------- HAZARD SUMMARY * Acrylonitrile can affect you when breathed in and by training concerning chemical hazards and controls. The federal passing through your skin. OSHA Hazard Communication Standard 29 CFR 1910.1200, * Acrylonitrile is a CARCINOGEN--HANDLE WITH requires private employers to provide similar training and EXTREME CAUTION. information to their employees. * Acrylonitrile should be handled as a TERATOGEN-- WITH EXTREME CAUTION. * Exposure to hazardous substances should be routinely * Skin contact can cause severe irritation and blistering. evaluated. This may include collecting personal and area air * Exposure to Acrylonitrile can irritate the eyes, nose, and samples. You can obtain copies of sampling results from throat. your employer. You have a legal right to this information * Breathing Acrylonitrile can irritate the lungs causing under the OSHA Standard 29 CFR 1910.1020. coughing and/or shortness of breath. Higher exposures can * If you think you are experiencing any work-related health cause a build-up of fluid in the lungs (pulmonary edema), a problems, see a doctor trained to recognize occupational medical emergency, with severe shortness of breath. diseases. Take this Fact Sheet with you. * Exposure to Acrylonitrile can cause weakness, headache, * ODOR THRESHOLD = 1.6 ppm. dizziness, confusion, nausea, vomiting, and can lead to * The range of accepted odor threshold values is quite broad. death. Caution should be used in relying on odor alone as a * Repeated exposure can irritate the nose causing discharge, warning of potentially hazardous exposures. -

Polymer Effect on Molecular Recognition. Enhancement of Molecular-Shape Selectivity for Polycyclic Aromatic Hydrocarbons by Poly(Acrylonitrile)
Polymer Journal, Vol.34, No. 6, pp 437—442 (2002) Polymer Effect on Molecular Recognition. Enhancement of Molecular-Shape Selectivity for Polycyclic Aromatic Hydrocarbons by Poly(acrylonitrile) Makoto TAKAFUJI, Wei DONG, Yoshihiro GOTO, Toshihiko SAKURAI, ∗ † Shoji NAGAOKA, and Hirotaka IHARA Department of Applied Chemistry & Biochemistry, Faculty of Engineering, Kumamoto University, Kumamoto 860–8555, Japan ∗Kumamoto Industrial Research Institute, Kumamoto 862–0901, Japan (Received December 10, 2001; Accepted April 25, 2002) ABSTRACT: Poly(acrylonitrile) immobilized onto porous silica (Sil-ANn) was prepared to evaluate the effect of polymerization degree of poly(acrylonitrile) on selective interaction with polycyclic aromatic hydrocarbons. The HPLC using the packed column (Sil-ANn) and an aqueous solution as a mobile phase showed higher selectivity for structural isomers of polycyclic aromatic hydrocarbons compared with simply cyanopropylated silica (Sil-CN) and alkylated silica (Sil-C4). It was considered that silica-supported poly(acrylonitrile) recognized molecular aromaticity of π-electron con- taining compounds rather than molecular hydrophobicity. Furthermore, similar results were obtained in the selectivity towards geometrical isomers such as trans- and cis-stilbenes or triphenylene and o-terphenyl and structural isomers such as o-, m-, p-terphenyls. Also the separation factors increased with an increase in polymerization degree of ANn. This paper discusses that polymeric structures enhance the selectivity. KEY WORDS Polymer Grafting -

Acrylonitrile
Acrylonitrile 107-13-1 Hazard Summary Exposure to acrylonitrile is primarily occupational: it is used in the manufacture of acrylic acid and modacrylic fibers. Acute (short-term) exposure of workers to acrylonitrile has been observed to cause mucous membrane irritation, headaches, dizziness, and nausea. No information is available on the reproductive or developmental effects of acrylonitrile in humans. Based on limited evidence in humans and evidence in rats, EPA has classified acrylonitrile as a probable human carcinogen (Group B1). Please Note: The main sources of information for this fact sheet are EPA's Integrated Risk Information System (IRIS) (4), which contains information on inhalation chronic toxicity of acrylonitrile and the RfC and the carcinogenic effects of acrylonitrile including the unit cancer risk for inhalation exposure, EPA's Health Effects Assessment for Acrylonitrile (6), and the Agency for Toxic Substances and Disease Registry's (ATSDR's) Toxicological Profile for Acrylonitrile (1). Uses Acrylonitrile is primarily used in the manufacture of acrylic and modacrylic fibers. It is also used as a raw material in the manufacture of plastics (acrylonitrile-butadiene-styrene and styrene-acrylonitrile resins), adiponitrile, acrylamide, and nitrile rubbers and barrier resins. (1,6) Sources and Potential Exposure Human exposure to acrylonitrile appears to be primarily occupational, via inhalation. (1) Acrylonitrile may be released to the ambient air during its manufacture and use. (1) Assessing Personal Exposure Acrylonitrile -

Toxicological Profile for Acetone Draft for Public Comment
ACETONE 1 Toxicological Profile for Acetone Draft for Public Comment July 2021 ***DRAFT FOR PUBLIC COMMENT*** ACETONE ii DISCLAIMER Use of trade names is for identification only and does not imply endorsement by the Agency for Toxic Substances and Disease Registry, the Public Health Service, or the U.S. Department of Health and Human Services. This information is distributed solely for the purpose of pre dissemination public comment under applicable information quality guidelines. It has not been formally disseminated by the Agency for Toxic Substances and Disease Registry. It does not represent and should not be construed to represent any agency determination or policy. ***DRAFT FOR PUBLIC COMMENT*** ACETONE iii FOREWORD This toxicological profile is prepared in accordance with guidelines developed by the Agency for Toxic Substances and Disease Registry (ATSDR) and the Environmental Protection Agency (EPA). The original guidelines were published in the Federal Register on April 17, 1987. Each profile will be revised and republished as necessary. The ATSDR toxicological profile succinctly characterizes the toxicologic and adverse health effects information for these toxic substances described therein. Each peer-reviewed profile identifies and reviews the key literature that describes a substance's toxicologic properties. Other pertinent literature is also presented, but is described in less detail than the key studies. The profile is not intended to be an exhaustive document; however, more comprehensive sources of specialty information are referenced. The focus of the profiles is on health and toxicologic information; therefore, each toxicological profile begins with a relevance to public health discussion which would allow a public health professional to make a real-time determination of whether the presence of a particular substance in the environment poses a potential threat to human health. -

Ethylene Oxide
ETHYLENE OXIDE Ethylene oxide was considered by previous IARC Working Groups in 1976, 1984, 1987, 1994, and 2007 (IARC, 1976, 1985, 1987, 1994, 2008). Since that time new data have become avail- able, which have been incorporated in this Monograph, and taken into consideration in the present evaluation. 1. Exposure Data 1.2 Uses Ethylene oxide is an important raw material 1.1 Identification of the agent used in the manufacture of chemical derivatives From IARC (2008), unless indicated otherwise that are the basis for major consumer goods in Chem. Abstr. Serv. Reg. No.: 75-21-8 virtually all industrialized countries. More than Chem. Abstr. Serv. Name: Oxirane half of the ethylene oxide produced worldwide Synonyms: 1,2-Epoxyethane is used in the manufacture of mono-ethylene glycol. Conversion of ethylene oxide to ethylene O glycols represents a major use for ethylene oxide in most regions: North America (65%), western Europe (44%), Japan (63%), China (68%), Other Asia (94%), and the Middle East (99%). Important C2H4O Relative molecular mass: 44.06 derivatives of ethylene oxide include di-ethylene Description: Colourless, flammable gas glycol, tri-ethylene glycol, poly(ethylene) (O’Neill, 2006) glycols, ethylene glycol ethers, ethanol-amines, Boiling-point: 10.6 °C (Lide, 2008) and ethoxylation products of fatty alcohols, Solubility: Soluble in water, acetone, fatty amines, alkyl phenols, cellulose and benzene, diethyl ether, and ethanol (Lide, poly(propylene) glycol (Occupational Safety and 2008) Health Administration, 2005; Devanney, 2010). Conversion factor: mg/m3 = 1.80 × ppm; A very small proportion (0.05%) of the annual calculated from: mg/m3 = (relative production of ethylene oxide is used directly in molecular weight/24.45) × ppm, assuming the gaseous form as a sterilizing agent, fumigant standard temperature (25 °C) and pressure and insecticide, either alone or in non-explo- (101.3 kPa). -

Naphthalene Photoinitiated Copolymerizafion of Unsaturated Monomers
Naphthalene photoinitiated copolymerizafion of unsaturated monomers J. BARTOŇ, I. ČAPEK, D. LATH, and E. LATHOVÁ Polymer Institute, Slovak Academy of Sciences, 809 34 Bratislava Received 18 February 1976 For the naphthalene photoinitiated copolymerization of acrylonitrile with styrene, methyl methacrylate, vinyl chloride, vinyl acetate, and isoprene the copolymerization parameters, number average molecular weights, and limiting viscosity numbers of copolymers were determined. It was found that copolyme rization rates increased by increasing the acrylonitrile concentration in feed. The results obtained point at the radical nature of copolymerization. При нафталином фотоинициированной сополимеризации акрилонитри- ла со стиролом, метилметакрилатом, винилхлоридом, винилацетатом и изопреном были установлены параметры сополимеризации, средний молекулярный вес и предельная характеристика вязкости сополимеров. Было установлено, что скорость сополимеризации увеличивается с повы шением концентрации акрилонитрила в затравке. Полученные результа ты указывают на радикальную сущность сополимеризации. In our papers [1—3] we have shown that aromatic hydrocarbons photoinitiated the polymerization of acrylonitrile. It was concluded that polymerization proceeds by a free radical mechanism. As initiating particles the ion radicals were con sidered. These initiating particles were formed in the subsequent reactions of the reaction product arising in the interaction between aromatic hydrocarbon in the first excited singlet state and acrylonitrile in the ground state. In this communication we report the results of study of copolymerization of acrylonitrile (AN) with methyl methacrylate (MMA), styrene (S), vinyl chloride (VC1), vinyl acetate (VAc), and isoprene (I) in the presence of naphthalene (N) acting as photoinitiator. Experimental Monomers, all commercial products were carefully purified by three times repeated rectification under reduced pressure of argon. Vinyl chloride was dried by passing it through silica gel column. -

Acrylonitrile
ACRYLONITRILE This substance was considered by previous Working Groups, in February 1978 (IARC, 1979) and March 1987 (IARC, 1987a). Since that time, new data have become available, and these have been incorporated into the monograph and taken into consi- deration in the present evaluation. 1. Exposure Data 1.1 Chemical and physical data 1.1.1 Nomenclature Chem. Abstr. Serv. Reg. No.: 107-13-1 Chem. Abstr. Name: 2-Propenenitrile Synonyms: AN; cyanoethylene; propenenitrile; VCN; vinyl cyanide 1.1.2 Structural and molecular formulae and relative molecular mass H2 CCHCN C3H3N Relative molecular mass: 53.06 1.1.3 Chemical and physical properties of the pure substance (a) Description: Colourless liquid (Verschueren, 1996) (b) Boiling-point: 77.3°C (Lide, 1995) (c) Melting-point: –83.5°C (Lide, 1995) 20 (d) Density: d4 0.8060 (Lide, 1995) (e) Spectroscopy data: Infrared, nuclear magnetic resonance and mass spectral data have been reported (Sadtler Research Laboratories, 1980; Brazdil, 1991) (f) Solubility: Soluble in water (7.35 mL/100 mL at 20°C); very soluble in acetone, benzene, diethyl ether and ethanol (Lide, 1995; Budavari, 1996) (g) Volatility: Vapour pressure, 13.3 kPa at 23°C; relative vapour density (air = 1), 1.83 (Verschueren, 1996) (h) Stability: Flash-point (open cup), 0°C; flammable; polymerizes spontaneously, particularly in the absence of oxygen, on exposure to visible light and in contact with concentrated alkali (Budavari, 1996) (i) Explosive limits: Lower, 3.05%; upper, 17.0% (Budavari, 1996) (j) Octanol/water partition coefficient (P): log P, 0.25 (Hansch et al., 1995) –43– 44 IARC MONOGRAPHS VOLUME 71 (k) Conversion factor: mg/m3 = 2.17 × ppm1 1.1.4 Technical products and impurities Acrylonitrile of 99.5–99.7% purity is available commercially, with the following specifications (ppm by weight, maximum): acidity (as acetic acid), 10; acetone, 75; ace- tonitrile, 300; acrolein, 1; hydrogen cyanide, 5; total iron, 0.1; oxazole, 10; peroxides (as hydrogen peroxide), 0.2; water, 0.5%; and nonvolatile matter, 100. -
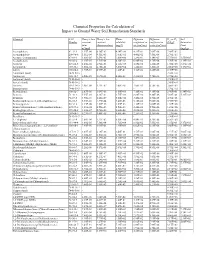
Chemical Properties Table
Chemical Properties for Calculation of Impact to Ground Water Soil Remediation Standards Chemical CAS Henry’s law Henry’s law Water Diffusion Diffusion Koc or Kd Soil Number constant constant solubility coefficient in coefficient in (L/kg)a Saturation (atm- (dimensionless) (mg/L) air (cm2/sec) water (cm2/sec) Limit m3/mol) (mg/kg) Acenaphthene 83-32-9 1.55E-04 6.36E-03 4.24E+00 4.21E-02 7.69E-06 7.08E+03 - Acenaphthylene 208-96-8 1.11E-04 4.51E-03 1.60E+01 4.40E-02 7.50E-06 2.76E+03 - Acetone (2-propanone) 67-64-1 3.88E-05 1.59E-03 1.00E+06 1.24E-01 1.14E-05 5.75E-01 1.55E+05 Acetophenone 98-86-2 1.10E-05 4.51E-04 6.10E+03 6.00E-02 8.70E-06 3.70E+01 1.39E+03 Acrolein 107-02-8 1.20E-04 4.92E-03 2.10E+05 1.05E-01 1.20E-05 1.00E+00 3.27E+04 Acrylonitrile 107-13-1 1.00E-04 4.10E-03 7.40E+04 1.22E-01 1.30E-05 2.00E+00 1.17E+04 Aldrin 309-00-2 1.70E-04 6.97E-03 1.80E-01 1.32E-02 4.86E-06 2.45E+06 - Aluminum (total) 7429-90-5 - - - - - 1.50E+03 - Anthracene 120-12-7 6.50E-05 2.67E-03 4.34E-02 3.24E-02 7.74E-06 2.95E+04 - Antimony (total) 7440-36-0 - - - - - 4.50E+01 - Arsenic (total) 7440-38-2 - - - - - 2.60E+01 - Atrazine 1912-24-9 2.96E-09 1.21E-07 7.00E+01 2.60E-02 6.70E-06 3.60E+02 - Barium (total) 7440-39-3 - - - - - 1.70E+01 - Benzaldehyde 100-52-7 2.67E-05 1.09E-03 3.00E+03 7.30E-02 9.10E-06 2.90E+01 6.34E+02 Benzene 71-43-2 5.55E-03 2.28E-01 1.75E+03 8.80E-02 9.80E-06 5.89E+01 5.22E+02 Benzidine 92-87-5 3.90E-11 1.60E-09 5.00E+02 3.40E-02 1.50E-05 4.70E+01 - Benzo(a)anthracene (1,2-Benzanthracene) 56-55-3 3.35E-06 1.37E-04 9.40E-03 5.10E-02 -

Acrylonitrile Safe Storage and Handling Guide Table of Contents
Acrylonitrile Safe Storage and Handling Guide Table of Contents Introduction 1.0 Physical, chemical and thermodynamic properties 2.0 Storage 3.0 Transfer and transport 4.0 Fire safety 5.0 Occupational safety and health 6.0 Medical response and first aid 7.0 Emergency response and environmental protection 8.0 Section 1: Introduction This booklet was prepared for individuals who handle or • Since pure Acrylonitrile may self-polymerize, causing rapid may come in contact with Acrylonitrile. It is a compilation build-up of pressure and resulting in an explosion hazard, of practical, understandable information designed to guide Innovene adds inhibitor prior to shipment. This additive the reader in responsible handling of Acrylonitrile, and to inhibits self-polymerization, but does not eliminate all answer commonly raised questions about Acrylonitrile possibility of polymerization. safety. Before handling, always consult a current Material • Fires involving Acrylonitrile can release toxic gases such as Safety Data Sheet (MSDS), available from INEOS, for hydrogen cyanide, oxides of nitrogen or carbon monoxide. information on the chemical. For more complete or detailed Its vapors can easily form explosive mixtures in air. Since information, call the following numbers in the U.S.: these vapors are heavier than air, they can also travel along the ground and be ignited by open flames or sparks which Emergency Notification Number 877-856-3682 are some distance from the site of release. Non-Emergency Information Numer 866-363-2454 • The vapors of Acrylonitrile, if inhaled at certain concentra- Information contained in this booklet is not intended to tions, can produce serious acute (short-term) toxicity, replace any legal requirements for Acrylonitrile handling including loss of consciousness or death. -

Pyrolysis and Combustion of Acetonitrile (Ch3cn)
OAK RIDGE ORNL/TM-2002/113 NATIONAL LABORATORY MANAGED BY UT-BATTELLE FOR THE DEPARTMENT OF ENERGY Pyrolysis and Combustion of Acetonitrile (CH3CN) Phillip F. Britt DOCUMENT AVAILABILITY Reports produced after January 1, 1996, are generally available free via the U.S. Department of Energy (DOE) Information Bridge. Web site http://www.osti.gov/bridge Reports produced before January 1, 1996, may be purchased by members of the public from the following source. National Technical Information Service 5285 Port Royal Road Springfield, VA 22161 Telephone 703-605-6000 (1-800-553-6847) TDD 703-487-4639 Fax 703-605-6900 E-mail [email protected] Web site http://www.ntis.gov/support/ordernowabout.htm Reports are available to DOE employees, DOE contractors, Energy Technology Data Exchange (ETDE) representatives, and International Nuclear Information System (INIS) representatives from the following source. Office of Scientific and Technical Information P.O. Box 62 Oak Ridge, TN 37831 Telephone 865-576-8401 Fax 865-576-5728 E-mail [email protected] Web site http://www.osti.gov/contact.html This report was prepared as an account of work sponsored by an agency of the United States Government. Neither the United States Government nor any agency thereof, nor any of their employees, makes any warranty, express or implied, or assumes any legal liability or responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process, or service by trade name, trademark, manufacturer, or otherwise, does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States Government or any agency thereof. -

Molecular Interactions of Acrylonitrile and Methylmethacrylate in Methanol, Cyclohexane & P-Dioxane
Available online a t www.pelagiaresearchlibrary.com Pelagia Research Library Advances in Applied Science Research, 2011, 2 (4):338-347 Molecular Interactions of Acrylonitrile and Methylmethacrylate in Methanol, Cyclohexane & P-dioxane 1V. D. Bhandakkar, 2G. R. Bedare, 3V. D. Muley and 4B. M. Suryavanshi, 1Department of Electronics, Anand Niketan College, Warora Dist– Chandrapur (MS), India 2Department of Physics, N. S. Sci. & Arts College, Bhadrawati Dist– Chandrapur (MS), India 3Department of Physics, J. M. Patel College, Bhandara Dist– Bhandara (MS), India 4Department of Physics, Institute of Science, Nagpur (MS), India ______________________________________________________________________________ ABSTRACT The Velocity of ultrasonic waves (10 MHz) for different compositions of mixtures of methanol, Cyclohexane and p-dioxane with acrylonitrile and methylmethacrylate has been measured in the 0 E E temperature range 10 - 40 C. The excess compressibility βa and excess molar volume V parameters have been discussed in the light of inter-molecular AB interactions and resulting E E disorder in these mixtures. It is observed that excess parameters βa and V can be used for classification of all types of molecular interactions in binary liquid mixtures. Keywords: Ultrasonic waves, Binary liquid mixtures, Methanol, Cyclohexane, p-dioxane, Acrylonitrile, Methylmethacrylate, Excess compressibility, Excess molar Volume. ______________________________________________________________________________ INTRODUCTION Ultrasonic parameters are being used extensively to study molecular interaction in pure liquid [1- 4] binary and ternary liquid mixtures [5,6] , and Ionic interactions in single mixed salt solution [5-7] . A departure from linearity in the velocity versus composition behaviour in liquid mixtures is taken as an indication of the existence of interaction between the different species. However, it is seen that a representation in terms of the observed parameters, such as velocity of ultrasonic waves, has a limited utility.