The Global Birth Defects Description and Coding (GBDDC) App
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Educational Paper Ciliopathies
Eur J Pediatr (2012) 171:1285–1300 DOI 10.1007/s00431-011-1553-z REVIEW Educational paper Ciliopathies Carsten Bergmann Received: 11 June 2011 /Accepted: 3 August 2011 /Published online: 7 September 2011 # The Author(s) 2011. This article is published with open access at Springerlink.com Abstract Cilia are antenna-like organelles found on the (NPHP) . Ivemark syndrome . Meckel syndrome (MKS) . surface of most cells. They transduce molecular signals Joubert syndrome (JBTS) . Bardet–Biedl syndrome (BBS) . and facilitate interactions between cells and their Alstrom syndrome . Short-rib polydactyly syndromes . environment. Ciliary dysfunction has been shown to Jeune syndrome (ATD) . Ellis-van Crefeld syndrome (EVC) . underlie a broad range of overlapping, clinically and Sensenbrenner syndrome . Primary ciliary dyskinesia genetically heterogeneous phenotypes, collectively (Kartagener syndrome) . von Hippel-Lindau (VHL) . termed ciliopathies. Literally, all organs can be affected. Tuberous sclerosis (TSC) . Oligogenic inheritance . Modifier. Frequent cilia-related manifestations are (poly)cystic Mutational load kidney disease, retinal degeneration, situs inversus, cardiac defects, polydactyly, other skeletal abnormalities, and defects of the central and peripheral nervous Introduction system, occurring either isolated or as part of syn- dromes. Characterization of ciliopathies and the decisive Defective cellular organelles such as mitochondria, perox- role of primary cilia in signal transduction and cell isomes, and lysosomes are well-known -

Potter Syndrome: a Case Study
Case Report Clinical Case Reports International Published: 22 Sep, 2017 Potter Syndrome: A Case Study Konstantinidou P1,2, Chatzifotiou E1,2, Nikolaou A1,2, Moumou G1,2, Karakasi MV2, Pavlidis P2 and Anestakis D1* 1Laboratory of Forensic and Toxicology, Department of Histopathology, Aristotle University of Thessaloniki, Greece 2Laboratory of Forensic Sciences, Department of Histopathology, Democritus University of Thrace School, Greece Abstract Potter syndrome (PS) is a term used to describe a typical physical appearance, which is the result of dramatically decreased amniotic fluid volume secondary to renal diseases such as bilateral renal agenesis (BRA). Other causes are abstraction of the urinary tract, autosomal recessive polycystic kidney disease (ARPKD), autosomal dominant polycystic kidney disease (ADPKD) and renal hypoplasia. In 1946, Edith Potter characterized this prenatal renal failure/renal agenesis and the resulting physical characteristics of the fetus/infant that result from oligohydramnios as well as the complete absence of amniotic fluid (anhydramnios). Oligohydramnios and anhydramnios can also be due to the result of leakage of amniotic fluid from rupturing of the amniotic membranes. The case reported concerns of stillborn boy with Potter syndrome. Keywords: Potter syndrome; Polycystic kidney disease; Oligohydramnios sequence Introduction Potter syndrome is not technically a ‘syndrome’ as it does not collectively present with the same telltale characteristics and symptoms in every case. It is technically a ‘sequence’, or chain of events - that may have different beginnings, but ends with the same conclusion. Below are the different ways that Potter syndrome (A.K.A Potter sequence) can begin due to various causes of renal failure. They gave numbers to differentiate the different forms, but this system had not caught in the medical and scientific communities [1]. -

Poland Syndrome with Atypical Malformations Associated to a De Novo 1.5 Mb Xp22.31 Duplication
Short Communication Poland Syndrome with Atypical Malformations Associated to a de novo 1.5 Mb Xp22.31 Duplication Carmela R. Massimino1 Pierluigi Smilari1 Filippo Greco1 Silvia Marino2 Davide Vecchio3 Andrea Bartuli3 Pasquale Parisi4 Sung Y. Cho5 Piero Pavone1,5 1 Department of Clinical and Experimental Medicine, Section of Pediatrics Address for correspondence Piero Pavone, MD, PhD, Department of and Child Neuropsychiatry, University of Catania, Catania, CT, Italy Pediatrics, AOU Policlinico-Vittorio Emanuele, University of Catania, 2 University-Hospital “Policlinico-Vittorio Emanuele,” University of Via S. Sofia 78, 95123 Catania, CT, Italy (e-mail: [email protected]). Catania, Catania, CT, Italy 3 Rare Disease and Medical Genetics, Academic Department of Pediatrics, Bambino Gesù Children’s Hospital, Rome, Italy 4 Child Neurology, Chair of Pediatrics, NESMOS Department, Faculty of Medicine & Psychology, Sapienza University, c/o Sant’ Andrea Hospital, Rome, Italy 5 Department of Pediatrics, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Republic of Korea Neuropediatrics Abstract Poland’s syndrome (PS; OMIM 173800) is a rare congenital syndrome which consists of absence or hypoplasia of the pectoralis muscle. Other features can be variably associated, including rib defects. On the affected side other features (such as of breast and nipple anomalies, lack of subcutaneous tissue and skin annexes, hand anomalies, visceral, and vertebral malformation) have been variably documented. To date, association of PS with central nervous system malformation has been rarely reported remaining poorly understood and characterized. We report a left-sided PS patient Keywords carrying a de novo 1.5 Mb Xp22.31 duplication diagnosed in addiction to strabismus, ► Poland’ssyndrome optic nerves and chiasm hypoplasia, corpus callosum abnormalities, ectopic neurohy- ► hypoplasic optic pophysis, pyelic ectasia, and neurodevelopmental delay. -

Potter Syndrome, a Rare Entity with High Recurrence Risk in Women with Renal Malformations – Case Report and a Review of the Literature
CASE PRESENTATIONS Ref: Ro J Pediatr. 2017;66(2) DOI: 10.37897/RJP.2017.2.9 POTTER SYNDROME, A RARE ENTITY WITH HIGH RECURRENCE RISK IN WOMEN WITH RENAL MALFORMATIONS – CASE REPORT AND A REVIEW OF THE LITERATURE George Rolea1, Claudiu Marginean1, Vladut Stefan Sasaran2, Cristian Dan Marginean2, Lorena Elena Melit3 1Obstetrics and Gynecology Clinic 1, Tirgu Mures 2University of Medicine and Pharmacy, Tirgu Mures 3Pediatrics Clinic 1, Tirgu Mures ABSTRACT Potter syndrome represents an association between a specific phenotype and pulmonary hypoplasia as a result of oligohydramnios that can appear in different pathological conditions. Thus, Potter syndrome type 1 or auto- somal recessive polycystic renal disease is a relatively rare pathology and with poor prognosis when it is diag- nosed during the intrauterine life. We present the case of a 24-year-old female with an evolving pregnancy, 22/23 gestational weeks, in which the fetal ultrasound revealed oligohydramnios, polycystic renal dysplasia and pulmonary hypoplasia. The personal pathological history revealed the fact that 2 years before this pregnancy, the patient presented a therapeutic abortion at 16 gestational weeks for the same reasons. The maternal ultra- sound showed unilateral maternal renal agenesis. Due to the fact that the identified fetal malformation was in- compatible with life, we decided to induce the therapeutic abortion. The particularity of the case consists in di- agnosing Potter syndrome in two successive pregnancies in a 24-year-old female, without any significant -

A Narrative Review of Poland's Syndrome
Review Article A narrative review of Poland’s syndrome: theories of its genesis, evolution and its diagnosis and treatment Eman Awadh Abduladheem Hashim1,2^, Bin Huey Quek1,3,4^, Suresh Chandran1,3,4,5^ 1Department of Neonatology, KK Women’s and Children’s Hospital, Singapore, Singapore; 2Department of Neonatology, Salmanya Medical Complex, Manama, Kingdom of Bahrain; 3Department of Neonatology, Duke-NUS Medical School, Singapore, Singapore; 4Department of Neonatology, NUS Yong Loo Lin School of Medicine, Singapore, Singapore; 5Department of Neonatology, NTU Lee Kong Chian School of Medicine, Singapore, Singapore Contributions: (I) Conception and design: EAA Hashim, S Chandran; (II) Administrative support: S Chandran, BH Quek; (III) Provision of study materials: EAA Hashim, S Chandran; (IV) Collection and assembly: All authors; (V) Data analysis and interpretation: BH Quek, S Chandran; (VI) Manuscript writing: All authors; (VII) Final approval of manuscript: All authors. Correspondence to: A/Prof. Suresh Chandran. Senior Consultant, Department of Neonatology, KK Women’s and Children’s Hospital, Singapore 229899, Singapore. Email: [email protected]. Abstract: Poland’s syndrome (PS) is a rare musculoskeletal congenital anomaly with a wide spectrum of presentations. It is typically characterized by hypoplasia or aplasia of pectoral muscles, mammary hypoplasia and variably associated ipsilateral limb anomalies. Limb defects can vary in severity, ranging from syndactyly to phocomelia. Most cases are sporadic but familial cases with intrafamilial variability have been reported. Several theories have been proposed regarding the genesis of PS. Vascular disruption theory, “the subclavian artery supply disruption sequence” (SASDS) remains the most accepted pathogenic mechanism. Clinical presentations can vary in severity from syndactyly to phocomelia in the limbs and in the thorax, rib defects to severe chest wall anomalies with impaired lung function. -

Familial Poland Anomaly
J Med Genet: first published as 10.1136/jmg.19.4.293 on 1 August 1982. Downloaded from Journal ofMedical Genetics, 1982, 19, 293-296 Familial Poland anomaly T J DAVID From the Department of Child Health, University of Manchester, Booth Hall Children's Hospital, Manchester SUMMARY The Poland anomaly is usually a non-genetic malformation syndrome. This paper reports two second cousins who both had a typical left sided Poland anomaly, and this constitutes the first recorded case of this condition affecting more than one member of a family. Despite this, for the purposes of genetic counselling, the Poland anomaly can be regarded as a sporadic condition with an extremely low recurrence risk. The Poland anomaly comprises congenital unilateral slightly reduced. The hands were normal. Another absence of part of the pectoralis major muscle in son (Greif himself) said that his own left pectoralis combination with a widely varying spectrum of major was weaker than the right. "Although the ipsilateral upper limb defects.'-4 There are, in difference is obvious, the author still had to carry addition, patients with absence of the pectoralis out his military duties"! major in whom the upper limbs are normal, and Trosev and colleagues9 have been widely quoted as much confusion has been caused by the careless reporting familial cases of the Poland anomaly. labelling of this isolated defect as the Poland However, this is untrue. They described a mother anomaly. It is possible that the two disorders are and child with autosomal dominant radial sided part of a single spectrum, though this has never been upper limb defects. -

Angiotensin Converting Enzyme (ACE) Inhibitors
J Med Genet 1997;34:541-545 541 Congenital renal tubular dysplasia and skull ossification defects similar to teratogenic effects of J Med Genet: first published as 10.1136/jmg.34.7.541 on 1 July 1997. Downloaded from angiotensin converting enzyme (ACE) inhibitors Dhavendra Kumar, Gail Moss, Rob Primhak, Robert Coombs Abstract An apparently autosomal recessive syn- drome of congenital renal tubular dyspla- sia and skull ossification defects is described in five infants from two sepa- rate, consanguineous, Pakistani Muslim kindreds. The clinical, pathological, and Centre for Human Genetics, 117 radiological features are similar to the Manchester Road, phenotype associated with fetal exposure Sheffield S1O 5DN, UK to angiotensin converting enzyme (ACE) D Kumar inhibitors: intrauterine growth retarda- Department of tion, skull ossification defects, and fetal/ Paediatrics, Sheffield neonatal anuric renal failure associated Children's Hospital, with renal tubular dysplasia. There was no Sheffield, UK fetal exposure to ACE inhibitors in the G Moss affected infants. similarities R Primhak Phenotypic between these familial cases and those Neonatal Unit, Jessop associated with ACE inhibition suggest an Hospital for Women, abnormality of the "renin-angiotensin- Sheffield, UK aldosterone" system (RAS). It is postu- R Coombs lated that the molecular pathology in this Correspondence to: uncommon autosomal recessive proximal Dr Kumar. renal tubular dysgenesis could be related http://jmg.bmj.com/ to mutations of the Received 4 March 1996 gene systems govern- Revised version accepted for ing the RAS. publication 6 March 1997 (JMed Genet 1997;34:541-545) Figure 2 Skull radiograph ofIV.5,family 1: note poor ossification and wide skull sutures. -
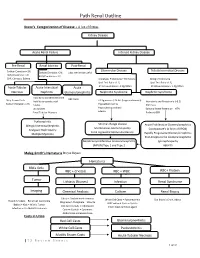
Path Renal Outline
Path Renal Outline Krane’s Categorization of Disease + A lot of Extras Kidney Disease Acute Renal Failure Intrinsic Kidney Disease Pre‐Renal Renal Intrinsic Post‐Renal Sodium Excretion <1% Glomerular Disease Tubulointerstitial Disease Sodium Excretion < 1% Sodium Excretion >2% Labs aren’t that useful BUN/Creatinine > 20 BUN/Creatinine < 10 CHF, Cirrhosis, Edema Urinalysis: Proteinuria + Hematuria Benign Proteinuria Spot Test Ratio >1.5, Spot Test Ratio <1.5, Acute Tubular Acute Interstitial Acute 24 Urine contains > 2.0g/24hrs 24 Urine contains < 1.0g/24hrs Necrosis Nephritis Glomerulonephritis Nephrotic Syndrome Nephritic Syndrome Inability to concentrate Urine RBC Casts Dirty Brown Casts Inability to secrete acid >3.5g protein / 24 hrs (huge proteinuria) Hematuria and Proteinuria (<3.5) Sodium Excretion >2% Edema Hypoalbuminemia RBC Casts Hypercholesterolemia Leukocytes Salt and Water Retention = HTN Focal Tubular Necrosis Edema Reduced GFR Pyelonephritis Minimal change disease Allergic Interstitial Nephritis Acute Proliferative Glomerulonephritis Membranous Glomerulopathy Analgesic Nephropathy Goodpasture’s (a form of RPGN) Focal segmental Glomerulosclerosis Rapidly Progressive Glomerulonephritis Multiple Myeloma Post‐Streptococcal Glomerulonephritis Membranoproliferative Glomerulonephritis IgA nephropathy (MPGN) Type 1 and Type 2 Alport’s Meleg‐Smith’s Hematuria Break Down Hematuria RBCs Only RBC + Crystals RBC + WBC RBC+ Protein Tumor Lithiasis (Stones) Infection Renal Syndrome Imaging Chemical Analysis Culture Renal Biopsy Calcium -
Spina Bifida & Certain Birth Defects
Spina Bifida & Certain Birth Defects Spina Bifida Benefits Eligibility. (38 U.S.C. 1805) There are three basic eligibility requirements: 1. The parent(s) of a spina bifida child-claimant must have performed active military, naval, or air service in the Republic of Vietnam between January 9, 1962 and May 7, 1975. 2. The child must be the natural child of the Vietnam veteran, regardless of age or marital status, who was conceived after the date on which the veteran first entered the Republic of Vietnam. The term “natural child” means a biological child and excludes the notion of deriving entitlement from adoptive parents. Only a biological parent of an adopted child could make the child eligible. 3. Spina Bifida benefits are payable for all types of spina bifida except spina bifida occulta. Private physicians, government or private institution examination reports may establish the diagnosis. Effective Date Level I Level II Level III 12/1/2003 $237 $821 $1,402 Children of Women Vietnam Veterans Born with Certain Birth Defects (38 U.S.C. 1815) Who is eligible for the Children of Women Vietnam Veterans monthly allowance? Under Public Law 106-419, children born to women Vietnam veterans may be eligible for a monthly monetary allowance if they suffer from certain covered birth defects. Children must have been conceived after the date on which the veteran first entered the Republic of Vietnam during the period beginning on February 28, 1961, and ending on May 7, 1975. (Spina Bifida however, is covered under the VA’s Spina Bifida Program.) VA identifies the birth defects as those that are associated with the service of the mother in Vietnam and resulted in permanent physical or mental disability. -

A Framework for the Evaluation of Patients with Congenital Facial Weakness Bryn D
Webb et al. Orphanet J Rare Dis (2021) 16:158 https://doi.org/10.1186/s13023-021-01736-1 REVIEW Open Access A framework for the evaluation of patients with congenital facial weakness Bryn D. Webb1,2* , Irini Manoli3, Elizabeth C. Engle4,5,6 and Ethylin W. Jabs1,2 Abstract There is a broad diferential for patients presenting with congenital facial weakness, and initial misdiagnosis unfortu- nately is common for this phenotypic presentation. Here we present a framework to guide evaluation of patients with congenital facial weakness disorders to enable accurate diagnosis. The core categories of causes of congenital facial weakness include: neurogenic, neuromuscular junction, myopathic, and other. This diagnostic algorithm is presented, and physical exam considerations, additional follow-up studies and/or consultations, and appropriate genetic testing are discussed in detail. This framework should enable clinical geneticists, neurologists, and other rare disease special- ists to feel prepared when encountering this patient population and guide diagnosis, genetic counseling, and clinical care. Keywords: Congenital facial weakness, Facial paralysis, Clinical genetics, Clinical characterization Clinical characteristics: congenital facial weakness CFW may be unilateral or bilateral and may be partial Congenital facial weakness (CFW) refers to decreased or complete (Fig. 2). Complete CFW refers to complete facial movement present at birth secondary to impaired absence of facial movement in all four quadrants of the function of facial musculature. CFW may be second- face (right upper quadrant, right lower quadrant, left ary to a defect in the motor nucleus of the facial nerve upper quadrant, and left lower quadrant). Patients with or the facial nerve itself (cranial nerve 7; CN7) (neuro- complete absence of facial movement on the left side genic), a defect at the neuromuscular junction, an inher- of the face may be described as having unilateral (left) ent muscular problem (myopathic), or other unknown or complete CFW. -

Meckel–Gruber Syndrome: an Update on Diagnosis, Clinical Management, and Research Advances
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by White Rose Research Online MINI REVIEW published: 20 November 2017 doi: 10.3389/fped.2017.00244 Meckel–Gruber Syndrome: An Update on Diagnosis, Clinical Management, and Research Advances Verity Hartill1,2, Katarzyna Szymanska2, Saghira Malik Sharif1, Gabrielle Wheway3 and Colin A. Johnson2* 1 Department of Clinical Genetics, Yorkshire Regional Genetics Service, Leeds Teaching Hospitals NHS Trust, Leeds, United Kingdom, 2 Leeds Institute of Biomedical and Clinical Sciences, University of Leeds, Leeds, United Kingdom, 3 Faculty of Health and Applied Sciences, Department of Applied Sciences, UWE Bristol, Bristol, United Kingdom Meckel–Gruber syndrome (MKS) is a lethal autosomal recessive congenital anomaly syndrome caused by mutations in genes encoding proteins that are structural or func- tional components of the primary cilium. Conditions that are caused by mutations in ciliary genes are collectively termed the ciliopathies, and MKS represents the most severe condition in this group of disorders. The primary cilium is a microtubule-based organelle, projecting from the apical surface of vertebrate cells. It acts as an “antenna” Edited by: that receives and transduces chemosensory and mechanosensory signals, but also Miriam Schmidts, regulates diverse signaling pathways, such as Wnt and Shh, that have important roles Radboud University Nijmegen, Netherlands during embryonic development. Most MKS proteins localize to a distinct ciliary com- Reviewed by: partment called the transition zone (TZ) that regulates the trafficking of cargo proteins Julia Hoefele, or lipids. In this review, we provide an up-to-date summary of MKS clinical features, Technische Universität München, Germany molecular genetics, and clinical diagnosis. -

16 the Kidney J
16 The Kidney J. Charles Jennette FPO FPO FPO FPO CONGENITAL ANOMALIES IgA Nephropathy (Berger Disease) Renal Agenesis Anti-Glomerular Basement Membrane Ectopic Kidney Glomerulonephritis Horseshoe Kidney ANCA Glomerulonephritis Renal Dysplasia VASCULAR DISEASES CONGENITAL POLYCYSTIC KIDNEY DISEASES Renal Vasculitis Autosomal Dominant Polycystic Kidney Disease Hypertensive Nephrosclerosis (Benign Nephrosclerosis) (ADPKD) Malignant Hypertensive Nephropathy Autosomal Recessive Polycystic Kidney Disease Renovascular Hypertension (ARPKD) Thrombotic Microangiopathy Nephronophthisis–Medullary Cystic Disease Cortical Necrosis ACQUIRED CYSTIC KIDNEY DISEASE DISEASES OF TUBULES AND INTERSTITIUM GLOMERULAR DISEASES Acute Tubular Necrosis (ATN) Nephrotic Syndrome Pyelonephritis Nephritic Syndrome Analgesic Nephropathy Glomerular Inflammation and Immune Mechanisms Drug-Induced (Hypersensitivity) Acute Tubulointerstitial Minimal-Change Glomerulopathy Nephritis Focal Segmental Glomerulosclerosis (FSGS) Light-Chain Cast Nephropathy Membranous Glomerulopathy Urate Nephropathy Diabetic Glomerulosclerosis RENAL STONES (NEPHROLITHIASIS AND Amyloidosis UROLITHIASIS) Hereditary Nephritis (Alport Syndrome) OBSTRUCTIVE UROPATHY AND HYDRONEPHROSIS Thin Glomerular Basement Membrane Nephropathy RENAL TRANSPLANTATION Acute Postinfectious Glomerulonephritis MALIGNANT TUMORS OF THE KIDNEY Type I Membranoproliferative Glomerulonephritis Wilms’ Tumor (Nephroblastoma) Type II Membranoproliferative Glomerulonephritis Renal Cell Carcinoma (RCC) (Dense Deposit Disease)