Preeclampsia, the Renin-Angiotensin-Aldosterone System and Beyond
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The (Pro)Renin Receptor: a New Kid in Town
The (Pro)Renin Receptor: A New Kid in Town Geneviève Nguyen, MD, PhD Summary: Renin inhibitors are now available in therapeutic doses and it is accepted that they decrease blood pressure as efficiently as the classic inhibitors of the renin-angiotensin system (RAS): angiotensin converting enzyme inhibitors and angiotensin II–receptor blockers (ARBs). One major issue will be to know how, beyond the normalization of blood pressure, renin inhibitors (RIs) will compare with angiotensin converting enzyme inhibitors and ARBs for their ability to protect the organs against the tissue damage associated with overactivation of the RAS. The mechanism(s) of tissue protection may involve the inhibition of a direct cellular effect of renin and prorenin mediated by the (pro)renin receptor ([P]RR). This review updates the recent findings on (P)RR; its role in hypertension, cardiac fibrosis, diabetic nephropathy, and retinopathy; and the effects of a putative (P)RR antagonist. Semin Nephrol 27:519-523 © 2007 Elsevier Inc. All rights reserved. Keywords: Renin-angiotensin system, renin, prorenin, and (pro)renin receptor he renin-angiotensin system (RAS) is be- THE (P)RR coming more and more complex. In 3 The (P)RR receptor is a 350-amino acid protein Tdecades, the classic intravascular system with no homology with any known protein. aimed at the generation of angiotensin II (Ang The primary structure analysis showed the ex- II), considered a unique biologically active pep- istence of the following: (1) a signal peptide, tide, has been enriched with new enzymes, which is indicative of a secreted protein; (2) a such as angiotensin converting enzyme 2 and large ectodomain responsible for renin and pro- chymase, and new receptors such as for angio- renin binding; (3) a single transmembrane do- tensin IV and for (pro)renin ([pro]renin refers main; and (4) a short cytoplasmic domain in- 1 to renin and prorenin collectively). -

Immunologic Effects of the Renin-Angiotensin System
BRIEF REVIEW www.jasn.org Immunologic Effects of the Renin-Angiotensin System Steven D. Crowley and Nathan P. Rudemiller Division of Nephrology, Department of Medicine, Durham Veterans Affairs and Duke University Medical Centers, Durham, North Carolina ABSTRACT Inappropriate activation of the renin-angiotensin system (RAS) exacerbates renal cell lineages that constitute the immune and vascular injury. Accordingly, treatment with global RAS antagonists attenuates system have the capacity to express RAS cardiovascular risk and slows the progression of proteinuric kidney disease. By re- components,11,12 and the effects of the ducing BP, RAS inhibitors limit secondary immune activation responding to hemo- RAS peptides and enzymes on inflamma- dynamic injury in the target organ. However, RAS activation in hematopoietic cells tory responses are quite diverse. How- has immunologic effects that diverge from those of RAS stimulation in the kidney ever, one recurring theme that emerges and vasculature. In preclinical studies, activating type 1 angiotensin (AT1) receptors from the work of several laboratories in- in T lymphocytes and myeloid cells blunts the polarization of these cells toward cluding our own is that activating AT1 proinflammatory phenotypes, protecting the kidney from hypertensive injury and receptors directly on hematopoietic cells fibrosis. These endogenous functions of immune AT1 receptors temper the patho- may provide a feedback, immunosup- genic actions of renal and vascular AT1 receptors during hypertension. By counter- pressive signal to temper or limit the acting the effects of AT1 receptor stimulation in the target organ, exogenous pathogenic actions of inappropriate administration of AT2 receptor agonists or angiotensin 1–7 analogs may similarly RAS activation in the kidney, vascula- limit inflammatory injury to the heart and kidney. -

Signal Transduction of the (Pro)Renin Receptor As a Novel Therapeutic Target for Preventing End-Organ Damage
Hypertension Research (2010) 33, 98–104 & 2010 The Japanese Society of Hypertension All rights reserved 0916-9636/10 $32.00 www.nature.com/hr REVIEW Signal transduction of the (pro)renin receptor as a novel therapeutic target for preventing end-organ damage Heiko Funke-Kaiser, Frank S Zollmann, Jan H Schefe and Thomas Unger The (pro)renin receptor ((P)RR) not only represents a novel component of the renin–angiotensin system but is also a promising novel drug target because of its crucial involvement in the pathogenesis of renal and cardiac end-organ damage. This review discusses the signal transduction of the (P)RR with its adapter protein promyelocytic zinc-finger protein, the impact of this receptor, especially on cardiovascular disease, and its putative interaction with renin inhibitors such as aliskiren. Furthermore, the increasing complexity regarding the cellular function of the (P)RR is addressed, which arises by the intimate link with proton pumps and the phosphatase PRL-1, as well as by the presence of different subcellular localizations and of a soluble isoform of the (P)RR. Finally, the rationale and strategy for the development of small-molecule antagonists of the (P)RR, called renin/ prorenin receptor blockers, are presented. Hypertension Research (2010) 33, 98–104; doi:10.1038/hr.2009.206; published online 11 December 2009 Keywords: promyelocytic zinc-finger protein; (pro)renin receptor; renin–angiotensin system; renin/prorenin receptor blockers; signal transduction INTRODUCTION SIGNAL TRANSDUCTION OF THE (P)RR Renin and prorenin -

Antihypertensive Agents Using ALZET Osmotic Pumps
ALZET® Bibliography References on the Administration of Antihypertensive Agents Using ALZET Osmotic Pumps 1. Atenolol Q7652: W. B. Zhao, et al. Stimulation of beta-adrenoceptors up-regulates cardiac expression of galectin-3 and BIM through the Hippo signalling pathway. British Journal of Pharmacology 2019;176(14):2465-2481 Agents: Isoproterenol; propranolol; carvedilol; atenolol; ICI-118551 Vehicle: saline; ascorbic acid, buffered; Route: SC; Species: Mice; Pump: 2001; Duration: 1 day; 2 days; 7 days; ALZET Comments: Dose ((ISO 0.6, 6, 20 mg/kg/d), (Prop 2 mg/kg/d), (Carv 2 mg/kg/d), (AT 2 mg/kg/d), (ICI 1 mg/kg/d)); saline with 0.4 mM ascorbic acid used; Controls were non-transgenic and received mp w/ vehicle; animal info (12-16 weeks, Male, (C57BL/6J, beta2-TG, Mst1-TG, or dnMst1-TG)); ICI-118551 is a beta2-antagonist with the structure (2R,3S)-1-[(7-methyl-2,3-dihydro-1H-inden-4-yl)oxy]-3-(propan-2-ylamino)butan-2-ol; cardiovascular; Minipumps were removed to allow for washout of ISO overnight prior to imaging; Q7241: M. N. Nguyen, et al. Mechanisms responsible for increased circulating levels of galectin-3 in cardiomyopathy and heart failure. Sci Rep 2018;8(1):8213 Agents: Isoproterenol, Atenolol, ICI-118551 Vehicle: Saline, ascorbic acid; Route: SC; Species: Mice; Pump: Not Stated; Duration: 48 Hours; ALZET Comments: Dose: ISO (2, 6 or 30 mg/kg/day; atenolol (2 mg/kg/day), ICI-118551 (1 mg/kg/day); 0.4 mM ascorbic used; animal info (12 14 week-old C57Bl/6 mice); cardiovascular; Q6161: C. -

(Pro)Renin Receptor–Mediated Signal Transduction and Tissue Renin
ORIGINAL ARTICLE (Pro)renin Receptor–Mediated Signal Transduction and Tissue Renin-Angiotensin System Contribute to Diabetes-Induced Retinal Inflammation Shingo Satofuka,1,2 Atsuhiro Ichihara,3 Norihiro Nagai,1,2 Kousuke Noda,1,2 Yoko Ozawa,1,2 Akiyoshi Fukamizu,4 Kazuo Tsubota,2 Hiroshi Itoh,3 Yuichi Oike,5 and Susumu Ishida1,2,6 OBJECTIVE—The term “receptor-associated prorenin sys- tem” (RAPS) refers to the pathogenic mechanisms whereby prorenin binding to its receptor dually activates the tissue ecause the renin-angiotensin system (RAS) plays renin-angiotensin system (RAS) and RAS-independent intra- an important role in the regulation of systemic cellular signaling via the receptor. The aim of the present blood pressure, RAS inhibitors including angio- study was to define the association of the RAPS with diabetes- tensin II type 1 receptor (AT1-R) blockers and induced retinal inflammation. B ACE inhibitors are safely and widely used in patients with RESEARCH DESIGN AND METHODS—Long-Evans rats, hypertension. In addition to strict control of blood glucose C57BL/6 mice, and angiotensin II type 1 receptor (AT1-R)- levels, tight blood pressure control with RAS inhibition deficient mice with streptozotocin-induced diabetes were treated has been shown to prevent the progression of diabetic with (pro)renin receptor blocker (PRRB). Retinal mRNA expres- retinopathy in the UK Prospective Diabetes Study (UKPDS) sion of prorenin and the (pro)renin receptor was examined by (1); however, diabetic patients are generally characterized quantitative RT-PCR. Leukocyte adhesion to the retinal vascula- by low renin and high prorenin levels in the plasma, ture was evaluated with a concanavalin A lectin perfusion– labeling technique. -
![Publication List Peer Reviewed Original Research Papers [44] WEN G, RINGSEIS R, EDER K](https://docslib.b-cdn.net/cover/5433/publication-list-peer-reviewed-original-research-papers-44-wen-g-ringseis-r-eder-k-2155433.webp)
Publication List Peer Reviewed Original Research Papers [44] WEN G, RINGSEIS R, EDER K
Publication list Peer reviewed original research papers [44] WEN G, RINGSEIS R, EDER K. Endoplasmic reticulum stress inhibits expression of genes involved in thyroid hormone synthesis and their key transcriptional regulators in FRTL-5 thyrocytes. PLoS One 2017, 12(11): e0187561. [43] WEN G, PACHNER L, GESSNER DK; EDER K, RINGSEIS R. Sterol Regulatory Element- Binding Proteins are Regulators of the Sodium/Iodide Symporter in Mammary Epithelial Cells. Journal of Dairy Science 2016, 99(11):9211-9226. [42] WEN G, EDER K, RINGSEIS R. Sterol regulatory element-binding proteins are transcriptional regulators of the thyroglobulin gene in thyroid cells. Biochimica et Biophysica Acta 2016, 1859(8):994-1003. [41] ZHOU X, RINGSEIS R, WEN G, EDER K. The pro-inflammatory cytokine tumor necrosis factor α stimulates expression of the carnitine transporter OCTN2 (novel organic cation transporter 2) and carnitine uptake via nuclear factor-κB in Madin-Darby bovine kidney cells. Journal Dairy Science 2015, 98(6):3840-8. [40] LUO H, ZHANG Y, GUO H, ZHANG L, LI X, RINGSEIS R, WEN G, HUI D, LIANG A, EDER K, HE D. Transcriptional regulation of the human, porcine and bovine OCTN2 gene by PPARα via a conserved PPRE located in intron 1. BMC Genetics 2014, 15(1):90. [39] ZHOU X, RINGSEIS R, WEN G, EDER K. The carnitine transporter OCTN2 and carnitine uptake in bovine kidney cells is regulated by peroxisome proliferator-activated receptor beta/delta. Acta Veterinaria Scandinavica 2014, 56:21. [38] RAUER C, RINGSEIS R, ROTHE S, WEN G, EDER K. Sterol regulatory element-binding proteins are regulators of the rat thyroid peroxidase gene in thyroid cells. -

Expression of Components of the Renin-Angiotensin System by Cancer Stem Cells in Renal Clear Cell Carcinoma
biomolecules Article Expression of Components of the Renin-Angiotensin System by Cancer Stem Cells in Renal Clear Cell Carcinoma Sam Siljee 1,† , Bridget Milne 1,†, Helen D. Brasch 1, Nicholas Bockett 1 , Josie Patel 1, Paul F. Davis 1, Andrew Kennedy-Smith 2, Tinte Itinteang 1 and Swee T. Tan 1,3,4,* 1 Gillies McIndoe Research Institute, Wellington 6242, New Zealand; [email protected] (S.S.); [email protected] (B.M.); [email protected] (H.D.B.); [email protected] (N.B.); [email protected] (J.P.); [email protected] (P.F.D.); [email protected] (T.I.) 2 Department of Urology, Wellington Regional Hospital, Wellington 6021, New Zealand; [email protected] 3 Wellington Regional Plastic, Maxillofacial and Burns Unit, Hutt Hospital, Lower Hutt 5010, New Zealand 4 Department of Surgery, The Royal Melbourne Hospital, The University of Melbourne, Melbourne, VIC 3010, Australia * Correspondence: [email protected]; Tel.: +64-(42)-820366 † Equal first authors. Abstract: This study investigated the expression of components of the renin-angiotensin system (RAS) by cancer stem cells (CSCs) we have recently demonstrated in renal clear cell carcinoma (RCCC). Fifteen RCCC tissue samples underwent immunohistochemical staining for components of the RAS: renin, pro-renin receptor (PRR), angiotensin-converting enzyme (ACE), angiotensin- converting enzyme 2 (ACE2), and angiotensin II receptor 2 (AT2R). Immunofluorescence co-staining or double immunohistochemical staining of these components of the RAS with stemness-associated Citation: Siljee, S.; Milne, B.; Brasch, markers OCT4 or KLF4 was performed on two of the samples. -
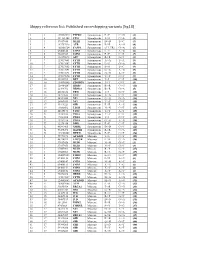
Skippy Reference List: Published Exon‐Skipping Variants (Hg18)
Skippy reference list: Published exon‐skipping variants (hg18) 1 1 196932540 PTPRC Synonymous P->P C->G (1) 2 2 211181402 CPS1 Synonymous S->S C->A (2) 3 3 37017458 MLH1 Synonymous D->D T->C (3) 4 3 143757998 ATR Synonymous E->E A->G (4) 5 4 185806724 CASP3 Synonymous (5’ UTR) G->A (5) 6 5 69408148 SMN1 Synonymous L->L A->G (6) 7 5 70283529 SMN2 Synonymous F->F C->T (7) 8 5 112198672 APC Synonymous R->R G->T (8) 9 7 117017661 CFTR Synonymous A->A T->A (9) 10 7 117017667 CFTR Synonymous L->L G->A (9) 11 7 117017682 CFTR Synonymous S->S T->C (9) 12 7 117017691 CFTR Synonymous G->G A->G (9) 13 7 117017691 CFTR Synonymous G->G A->T (9) 14 7 117017694 CFTR Synonymous Y->Y C->T (9) 15 10 42929995 RET Synonymous I->I C->T (10) 16 10 114190345 ZDHHC6 Synonymous T->T C->T (5) 17 11 118464207 HMBS Synonymous R->R C->G (11) 18 14 22444702 RBM23 Synonymous R->R G->A (5) 19 15 46516836 FBN1 Synonymous I->I C->T (12) 20 15 78251635 FAH Synonymous N->N C->T (13) 21 17 26551622 NF1 Synonymous Q->Q G->A (14) 22 17 26689883 NF1 Synonymous Y->Y C->T (15) 23 17 59349223 GH1 Synonymous E->E A->G (16) 24 19 11088602 LDLR Synonymous N->N C->T (17) 25 20 44185176 CD40 Synonymous T->T A->T (18) 26 X 19281185 PDHA Synonymous Y->Y C->T (19) 27 X 19281200 PDHA Synonymous I->I C->T (19) 28 X 19282574 PDHA Synonymous G->G A->G (20) 29 X 32276456 DMD Synonymous F->F C->T (21) 30 X 40341465 XMRE Synonymous D->D C->T (22) 31 X 53475492 HADH2 Synonymous R->R C->A (23) 32 X 133460368 HPRT1 Synonymous F->F C->T (24) 33 1 75971876 ACADM Missense T->I C->T (25) 34 2 48774922 -

X-Linked Mental Retardation
REVIEWS X-LINKED MENTAL RETARDATION H.-Hilger Ropers* and Ben C. J. Hamel‡ Abstract | Genetic factors have an important role in the aetiology of mental retardation. However, their contribution is often underestimated because in developed countries, severely affected patients are mainly sporadic cases and familial cases are rare. X-chromosomal mental retardation is the exception to this rule, and this is one of the reasons why research into the genetic and molecular causes of mental retardation has focused almost entirely on the X-chromosome. Here, we review the remarkable recent progress in this field, its promise for understanding neural function, learning and memory, and the implications of this research for health care. Mental retardation is one of the main reasons for refer- subject (by Chelly and Mandel7) was published. Pro- ral in paediatric, child-neurological and clinical genetic gress has been particularly spectacular for so-called practice. Often, however, despite extensive investiga- non-syndromic or ‘pure’ forms of XLMR, consisting of tions, an aetiological diagnosis cannot be made, leaving numerous disorders that are clinically indistinguishable families without accurate genetic counselling or repro- because cognitive impairment is their only manifestation. ductive options, such as prenatal diagnosis. The preva- In this review, we describe the strategies used to iden- lence of mental retardation in developed countries is tify all genes that have been implicated in XLMR so far, thought to be on the order of 2–3% (REF.1), although emphasizing the crucial importance of large cohorts of estimates vary widely, particularly for mild mental retar- well-characterized families. We provide a brief summary dation. -

Key Developments in Renin–Angiotensin–Aldosterone System
REVIEWS Key developments in renin–angiotensin– aldosterone system inhibition Bruno Sevá Pessôa, Nils van der Lubbe, Koen Verdonk, Anton J. M. Roks, Ewout J. Hoorn and A. H. Jan Danser Abstract | The renin–angiotensin–aldosterone system (RAAS) was initially thought to be fairly simple. However, this idea has been challenged following the development of RAAS blockers, including renin inhibitors, angiotensin-converting-enzyme (ACE) inhibitors, type 1 angiotensin II (AT1)-receptor blockers and mineralocorticoid-receptor antagonists. Consequently, new RAAS components and pathways that might contribute to the effectiveness of these drugs and/or their adverse effects have been identified. For example, an increase in renin levels during RAAS blockade might result in harmful effects via stimulation of the prorenin receptor (PRR), and prorenin—the inactive precursor of renin—might gain enzymatic activity on PRR binding. The increase in angiotensin II levels that occurs during AT1-receptor blockade might result in beneficial effects via stimulation of type 2 angiotensin II receptors. Moreover, angiotensin 1–7 levels increase during ACE inhibition and AT1-receptor blockade, resulting in Mas receptor activation and the induction of cardioprotective and renoprotective effects, including stimulation of tissue repair by stem cells. Finally, a role of angiotensin II in sodium and potassium handling in the distal nephron has been identified. This finding is likely to have important implications for understanding the effects of RAAS inhibition on whole body -

Serum and Glucocorticoid Regulated Kinase 1 in Sodium Homeostasis
International Journal of Molecular Sciences Review Serum and Glucocorticoid Regulated Kinase 1 in Sodium Homeostasis Yiyun Lou 1,2, Fan Zhang 1, Yuqin Luo 1, Liya Wang 1, Shisi Huang 1 and Fan Jin 1,3,* 1 Department of Reproductive Endocrinology, Women’s Hospital, School of Medicine, Zhejiang University, Hangzhou 310006, Zhejiang, China; [email protected] (Y.L.); [email protected] (F.Z.); [email protected] (Y.Lu.); [email protected] (L.W.); [email protected] (S.H.) 2 Department of Gynaecology, Hangzhou Hospital of Traditional Chinese Medicine, Hangzhou 310007, Zhejiang, China 3 Key Laboratory of Reproductive Genetics, National Ministry of Education (Zhejiang University), Women’s Reproductive Healthy Laboratory of Zhejiang Province, Hangzhou 310058, Zhejiang, China * Correspondence: [email protected]; Tel.: +86-571-8701-3891; Fax: +86-571-8706-1878 Academic Editor: Atsushi Matsuzawa Received: 15 June 2016; Accepted: 3 August 2016; Published: 10 August 2016 Abstract: The ubiquitously expressed serum and glucocorticoid regulated kinase 1 (SGK1) is tightly regulated by osmotic and hormonal signals, including glucocorticoids and mineralocorticoids. Recently, SGK1 has been implicated as a signal hub for the regulation of sodium transport. SGK1 modulates the activities of multiple ion channels and carriers, such as epithelial sodium channel (ENaC), voltage-gated sodium channel (Nav1.5), sodium hydrogen exchangers 1 and 3 (NHE1 and NHE3), sodium-chloride symporter (NCC), and sodium-potassium-chloride cotransporter 2 (NKCC2); as well as the sodium-potassium adenosine triphosphatase (Na+/K+-ATPase) and type A natriuretic peptide receptor (NPR-A). Accordingly, SGK1 is implicated in the physiology and pathophysiology of Na+ homeostasis. -

(Pro)Renin Receptor in Regulating Systemic Blood Pressure
HHS Public Access Author manuscript Author ManuscriptAuthor Manuscript Author Pharmacol Manuscript Author Ther. Author Manuscript Author manuscript; available in PMC 2017 August 01. Published in final edited form as: Pharmacol Ther. 2016 August ; 164: 126–134. doi:10.1016/j.pharmthera.2016.04.006. The critical role of the central nervous system (pro)renin receptor in regulating systemic blood pressure Quanbin Xu1, Dane D. Jensen1, Hua Peng2, and Yumei Feng1 1Departments of Pharmacology and Physiology & Cell Biology, Center for Cardiovascular Research, University of Nevada School of Medicine, Reno, NV, USA 2Department of Pediatrics, Union Hospital, Tongji Medical College, Huangzhong University of Sciences and Technology, Wuhan, China Abstract The systemic renin–angiotensin system (RAS) has long been recognized as a critically important system in blood pressure (BP) regulation. However, extensive evidence has shown that a majority of RAS components are also present in many tissues and play indispensable roles in BP regulation. Here, we review evidence that RAS components, notably including the newly identified (pro)renin receptor (PRR), are present in the brain and are essential for the central regulation of BP. Binding of the PRR to its ligand, prorenin or renin, increases BP and promotes progression of cardiovascular diseases in an angiotensin II-dependent and -independent manner, establishing the PRR a promising antihypertensive drug target. We also review the existing PRR blockers, including handle region peptide and PRO20, and propose a rationale for blocking prorenin/PRR activation as a therapeutic approach that does not affect the actions of the PRR in vacuolar H+- ATPase and development. Finally, we summarize categories of currently available antihypertensive drugs and consider future perspectives.